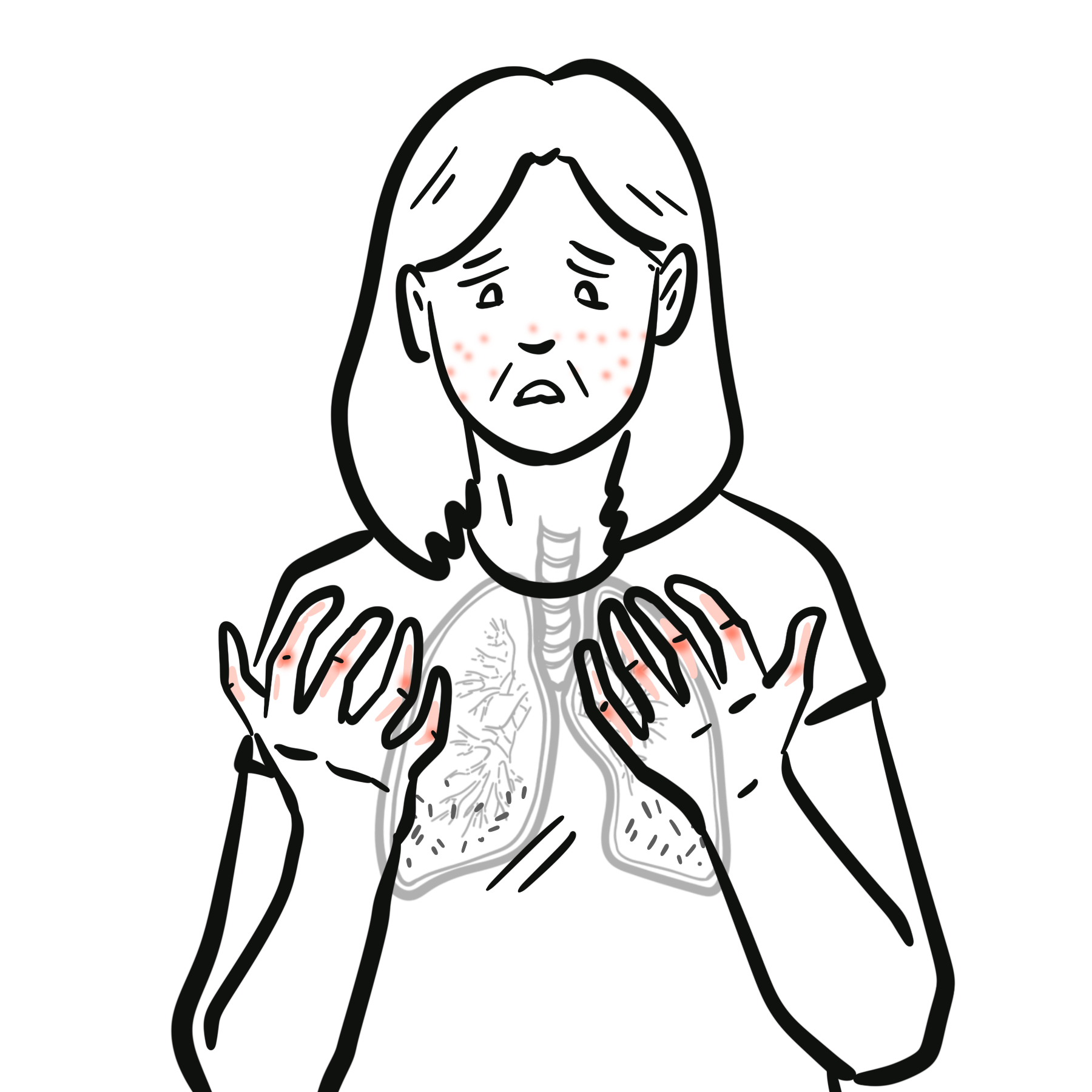
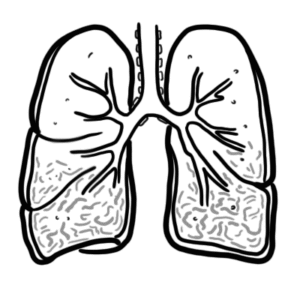
Systemic sclerosis–associated interstitial lung disease (SSc-ILD) is one of the most common and severe internal organ manifestations of systemic sclerosis (SSc), occurring in up to 50–60% of patients on HRCT, with clinically significant disease in ~25–30%. It is a major cause of SSc-related mortality. The most frequent histopathological pattern is nonspecific interstitial pneumonia (NSIP), followed by usual interstitial pneumonia (UIP). Risk factors include diffuse cutaneous SSc, anti-topoisomerase I (Scl-70) antibody positivity, male sex, African-American ethnicity, and early disease onset. The most important complication is progressive pulmonary fibrosis leading to respiratory failure and death.
Definition
Systemic sclerosis (SSc): Chronic autoimmune connective tissue disease characterized by vasculopathy, immune dysregulation, and fibrosis of skin and internal organs. Interstitial lung disease (ILD): Group of disorders involving inflammation and/or fibrosis of the lung interstitium. Nonspecific interstitial pneumonia (NSIP): ILD pattern with uniform interstitial inflammation and fibrosis, common in SSc. Forced Vital Capacity (FVC): Spirometric measure of lung volume, used to monitor ILD progression.
Anatomy and Physiology
Alveolar-capillary unit: Site of gas exchange; consists of alveolar epithelium, interstitial space, and capillary endothelium.
Type I pneumocytes: Thin cells covering most alveolar surface, specialized for gas exchange.
Type II pneumocytes: Produce surfactant, repair alveolar epithelium after injury.
Early detection and aggressive management improve survival.
References
Hoffmann-Vold AM et al. The identification and management of interstitial lung disease in systemic sclerosis: evidence-based European consensus statements. Lancet Rheumatol. 2020;2(2):e71–e83.
Distler O et al. Nintedanib for systemic sclerosis–associated interstitial lung disease. N Engl J Med. 2019;380:2518–2528.
Volkmann ER. Natural history of systemic sclerosis–related interstitial lung disease: how to identify a progressive fibrosing phenotype. J Scleroderma Relat Disord. 2020;5(2_suppl):31–40.
Goh NS et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med. 2008;177:1248–1254.
Bruni C et al. Predicting ILD in systemic sclerosis: the ILD-RISC model. BMC Pulm Med. 2025;25:3722.
Khanna D et al. 2013 ACR/EULAR classification criteria for systemic sclerosis. Arthritis Rheum. 2013;65:2737–2747.













Discussion