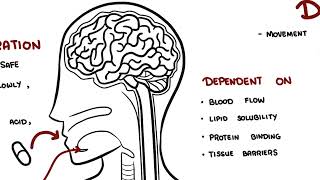
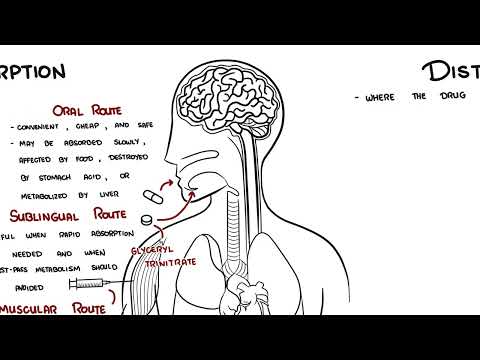
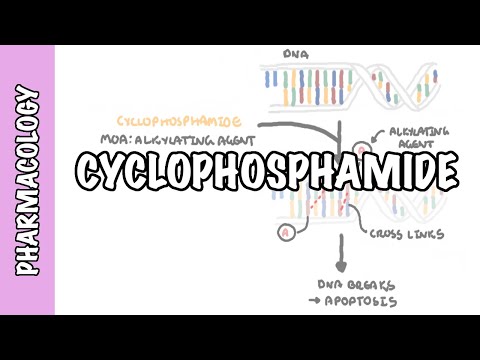
0:00 Pharmacokinetics is the study of what the body does to a drug. 0:09 It explains how a medication enters the body, moves through the bloodstream, 0:13 reaches the 0:14 tissues, is chemically changed and is eventually removed. 0:19 The four main stages of pharmacokinetics are remembered as ADME. 0:25 A is absorption, D is distribution, M is metabolism, and E is excretion. 0:32 Let's look at each of these briefly. 0:39 So absorption A is the movement of a drug from its site of administration into 0:45 the bloodstream. 0:47 For a drug to have a systemic effect, it usually needs to reach the circulation 0:53 first. 0:53 For example, when a patient swallows a tablet, the drug must dissolve in the 0:59 gastrointestinal 0:59 tract, pass across the gut wall, and enter the bloodstream. 1:04 Oral administration is common because it is convenient, cheap, and safe. 1:10 However, oral drugs may be absorbed slowly affected by food, destroyed by 1:15 stomach acid, 1:16 or metabolized before they actually reach the systemic circulation. 1:20 In contrast, intravenous drugs are injected directly into the bloodstream, so 1:26 they act 1:27 rapidly and have 100% bioavailability. 1:33 Bioavailability refers to the portion of a drug dose that reaches the systemic 1:38 circulation 1:39 unchanged. 1:41 Intravenous drugs have 100% bioavailability because the full dose enters the 1:48 blood directly. 1:49 Oral drugs often have lower bioavailability because some of the drugs may not 1:55 be absorbed 1:56 or may be metabolized before reaching the bloodstream, as shown in this diagram 2:02 . 2:03 For example, if a patient takes 100mg tablet and only 50mg reaches the 2:08 circulation unchanged, 2:10 the bioavailability is 50%. 2:14 A related concept is first passed metabolism. 2:17 This occurs when an oral drug is absorbed from the gut and travels through the 2:22 portal 2:22 vein to the liver before entering the systemic circulation. 2:27 The liver may metabolize part of the drug during this first pass, reducing the 2:34 amount 2:35 of active drug available. 2:38 This is why some drugs are not taken orally. 2:42 A classic example is glyceral trinitrate, which is often given sublingually for 2:49 angina 2:50 chest pain, so it can enter the blood quickly and avoid significant first pass 3:00 metabolism. 3:02 The next stage of pharmacokinetics is distribution. 3:05 So distribution is the movement of a drug from the bloodstream into tissues and 3:11 organs. 3:12 So once the drug enters the blood, it may remain in the plasma or move into 3:17 areas such 3:18 as the brain, liver, kidneys, muscle, fat, placenta, or breast milk. 3:27 Distribution depends on several factors including blood flow, lipid solubility, 3:34 protein binding 3:35 and tissue barriers, organs with high blood flow such as the brain, heart, 3:42 liver and kidneys 3:44 receive drugs quickly, lipid soluble drugs cross cell membranes more easily and 3:50 may enter 3:51 the brain, fat, tissue, placenta and breast milk. 3:59 Protein binding is also important, many drugs bind to plasma proteins, 4:05 especially albumin. 4:06 The bound drug usually acts as a reservoir, while the free unbound drug is the 4:11 active 4:12 form that can leave the bloodstream, bind to receptors, be metabolized and be 4:18 excreted. 4:19 Warfarin is a classic example of a highly bound drug. 4:24 If another medication displaces warfarin from albumin, the amount of free war 4:28 farin may 4:28 increase, raising the risk of bleeding. 4:33 Volume of distribution or VD describes how widely a drug distributes into body 4:39 tissues 4:40 compared with plasma. 4:42 It is a theoretical value, not a real anatomical volume. 4:47 A drug with a low VD mostly stays in the bloodstream, while a drug with a high 4:53 VD moves widely 4:54 into tissues. 4:56 For example, heparin mainly stays within the vascular compartment, while dig 5:01 oxin distributes 5:02 extensively into tissues. 5:09 The next stage of pharmacokinetics is metabolism. 5:14 Metabolism is the chemical modification of a drug by the body. 5:18 The liver is the main organ responsible. 5:23 Metabolism often makes drugs more water soluble, so they can be excreted more 5:28 easily, such 5:29 as in the urine. 5:31 However, metabolism actually does not simply mean break down a drug. 5:38 Metabolism can also be inactivating a drug, or activating a prodrug, or 5:44 creating an active 5:46 metabolite or produce a toxic metabolite. 5:50 Metabolism is an example of a prodrug, because it is partly converted into 5:54 morphine, which 5:55 contributes to its pain-relieving effect. 6:00 Drug metabolism is often divided into Phase 1 and Phase 2 reactions. 6:07 Phase 1 reactions include oxidation, reduction, and hydrolysis, often involving 6:13 cytochrome 6:14 P450 enzymes. 6:17 These two reactions involve conjugation, where another molecule is attached to 6:23 the drug to 6:24 make it more water soluble, and when it's water soluble, it can then be excret 6:29 ed more 6:30 easily in the urine or in the bile. 6:33 Drug interactions commonly occur when one drug induces or inhibits liver 6:39 enzymes. 6:40 Enzyme induces speed up metabolism, and may reduce drug levels, while enzyme 6:45 inhibitors 6:46 use low metabolism, and may increase drug levels and toxicity. 6:55 The last stage is excretion. 6:59 Excretion is a removal of drugs from the body. 7:03 The kidneys are the main organs responsible, although drugs can also be excret 7:07 ed through 7:08 bile, feces, lungs, sweat, saliva, and breast milk. 7:13 Bile excretion involves filtration of the drug, secretion, and reabsorption of 7:19 the drug. 7:21 If kidney function is reduced, drugs cleared by the kidneys can accumulate in 7:27 the body 7:28 and cause toxicity. 7:31 Gentamicin is an important example because it is really clear and can cause 7:36 kidney and 7:37 hearing toxicity if levels become too high. 7:42 On the topic of excretion, it's important to know two essential dosing concepts 7:48 , clearance 7:49 and half-life. 7:52 Clearance describes how efficiently the body removes a drug from the blood. 7:56 Half-life is the time it takes for the drug concentration to fall by 50%. 8:02 Half-life helps determine how often a drug should be taken, how long it takes 8:07 to reach 8:08 steady state, and how long it remains after stopping. 8:12 Any state occurs when the rate of drug administration equals the rate of 8:17 elimination, usually after 8:19 four to five half-lives. 8:22 Finally, pharmacokinetics varies between patients. 8:28 Age, pregnancy, kidney disease, liver disease, body composition, and drug 8:33 interactions can 8:34 all change drug handling. 8:36 This is why prescribing must be individualized. 8:40 So in summary, pharmacokinetics is essential because it links basic science to 8:45 safe clinical 8:46 use. 8:47 It explains drug route, dose, timing, monitoring, interactions, and toxicity. 8:53 Thank you for watching.