Hyper-IgE syndrome (HIES) is a rare primary immunodeficiency characterised by markedly elevated serum IgE, recurrent staphylococcal skin and lung infections, eczema, and connective tissue/ skeletal abnormalities. Most commonly caused by autosomal dominant mutations in STAT3, it results in impaired Th17 cell differentiation and defective neutrophil chemotaxis. Autosomal recessive forms (DOCK8, TYK2 mutations) present with more severe viral infections and malignancy risk. Estimated prevalence is <1 in 1,000,000.
Definition
IgE: Immunoglobulin involved in allergic responses and defence against parasites. Th17 cells: Subset of CD4+ T-cells that recruit neutrophils to fight fungi and extracellular bacteria. STAT3: Transcription factor required for Th17 development; its mutation is the main cause of AD-HIES. Cold abscess: Staphylococcal abscess lacking signs of inflammation (heat, erythema).
Hyper-IgE Triad: Recurrent staphylococcal skin abscesses, pneumonia with pneumatoceles and eczema.
Anatomy & Physiology
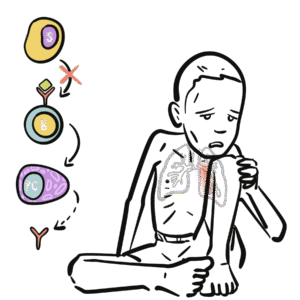
Normal pathway: Antigen → dendritic cell → naïve CD4+ T cell → Th17 cell differentiation via IL-6/IL-23 → neutrophil recruitment.
In HIES: Defective STAT3 or related genes → impaired Th17 differentiation → defective neutrophil chemotaxis and poor pathogen clearance.
Excessive IgE production contributes to atopy-like features and eosinophilia.
High IgE alone ≠ HIES (e.g., atopy), must have clinical triad of infections + eczema + IgE elevation.
Skin care: Anti-staphylococcal and anti-eczema management.
Immunotherapy: IVIG in recurrent/severe infections.
HSCT: Only curative in AR-HIES (DOCK8 deficiency); not effective in STAT3 HIES.
Supportive: Orthopaedic and dental care for skeletal/dental abnormalities.
AR (DOCK8) → consider HSCT; AD (STAT3) → supportive.
Complications & Prognosis
Recurrent severe infections → bronchiectasis, pneumatoceles, chronic lung disease.
Skeletal complications: scoliosis, fractures.
Malignancy risk (esp. lymphoma, SCC) in AR forms.
Prognosis: variable; supportive care has improved survival, but complications reduce quality of life.
Prognosis worse in AR-HIES due to severe viral infections and cancer risk.
References
Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357(16):1608–19.
Freeman AF, Holland SM. Clinical manifestations, etiology, and pathogenesis of the hyper-IgE syndromes. Pediatr Res. 2009;65(5 Pt 2):32R–37R.
Engelhardt KR, McGhee S, Winkler S, et al. Large deletions and point mutations involving DOCK8 in the autosomal recessive form of Hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124(6):1289–302.
Woellner C, Gertz EM, Schäffer AA, et al. Mutations in the signal transducer and activator of transcription 3 (STAT3) and diagnostic guidelines for hyper-IgE syndrome. J Allergy Clin Immunol. 2010;125(2):424–32.
Aydin SE, Kilic SS, Aytekin C, et al. DOCK8 deficiency: Clinical and immunological phenotype and treatment options – a review of 136 patients. J Clin Immunol. 2015;35(2):189–98.









Discussion