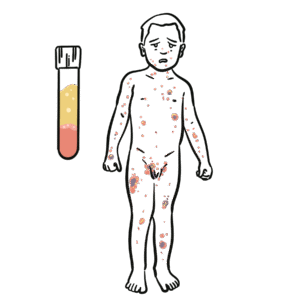
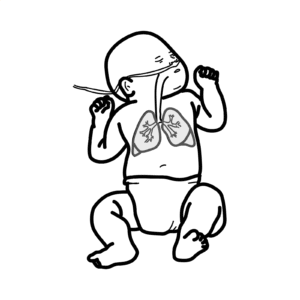
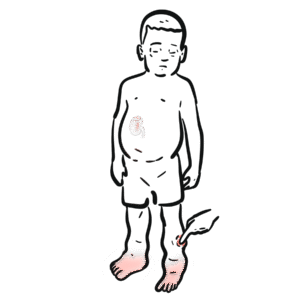
Severe combined immunodeficiency (SCID) is the most severe form of primary immunodeficiency, characterised by profound defects in both humoral (B-cell) and cellular (T-cell) immunity. It presents in infancy with recurrent, severe, and opportunistic infections, chronicdiarrhoea, and failure to thrive. Without curative therapy, most infants die within the first year of life. Incidence is ~1 in 50,000–100,000 live births. Newborn screening with T-cell receptor excision circles (TRECs) has enabled earlier detection.
Definition
Opportunistic infection: Infection by organisms that usually do not cause disease in immunocompetent hosts (e.g., Candida, Pneumocystis jirovecii). TRECs: T-cell receptor excision circles, used in newborn screening to detect impaired thymic T-cell production. X-linked SCID: Caused by mutations in the common γ-chain (IL2RG gene), the most common form. ADA deficiency: Adenosine deaminase deficiency; a metabolic cause of SCID.
Anatomy & Physiology
Normal lymphocyte development:
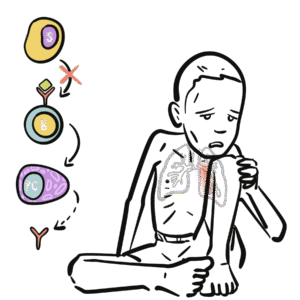
T cells mature in the thymus → mediate cellular immunity, help B cells.
B cells mature into plasma cells → produce immunoglobulins.
In SCID → T-cell development is severely impaired → defective cellular and humoral immunity.
NK cell numbers may also be reduced depending on subtype.
In SCID, defective T cells mean defective B-cell, even if B cells are present.
Aetiology
Genetic causes (major):
X-linked SCID (IL2RG mutation, defective common γ-chain).









Discussion