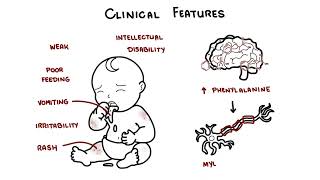
0:00 Phenylketouria, also known as PKU, is a rare genetic disorder that affects 0:10 approximately 0:11 6 per 100,000 births worldwide. 0:14 It's caused by an inborn error of metabolism, affecting the phenylalanine hyd 0:19 roxylase, or 0:20 PAH gene, which results in the absence or deficiency of phenylalanine hydroxyl 0:28 ase, PAH. 0:29 An enzyme responsible for breaking down the amino acid phenylalanine to tyros 0:37 ine. 0:38 Accumulation of phenylalanine is toxic to the brain and without treatment most 0:43 people develop 0:44 severe intellectual disability. 0:47 Ospion falling is a Norwegian biochemist that first described PKU in 1934. 0:54 Now let's talk about genetics. 1:01 So in biology normally people have a total of 46 chromosomes or 23 pairs, as 1:05 shown in 1:06 this diagram. 1:07 PKU is caused by variants in the PAH gene found on chromosome 12, specifically 1:15 chromosome 1:16 12Q23.2. 1:19 This just means a mutation in chromosome 12, essentially. 1:26 PKU is a genetic problem and is inherited in an autosomal recessive pattern. 1:37 So this means an individual must inherit two defective copies of the PAH gene, 1:45 of the phenylalanine 1:46 hydroxylase gene, to develop the disorder. 1:51 So they have to receive a defective copy, one from each parent. 1:57 As you can see in red, the two chromosomes have the red, which symbolizes the 2:02 mutation 2:03 in the PAH gene. 2:08 M stands for mutated gene. 2:12 If an individual inherits one normal gene and one disease causing gene variant, 2:17 they will 2:17 be a carrier of the disease and will usually not show symptoms. 2:22 In this image N means normal without a mutation and M again is a mutated gene. 2:33 So let's look at some examples and see how phenylketonuria, or PKU, is 2:40 inherited. 2:41 Let's look at an example where you have two carrier parents. 2:45 So both are carriers of the PKU mutation. 2:48 Each has one normal allele and one mutated allele, as shown in this diagram, 2:54 therefore 2:54 both are NM and NM. 2:58 So the possible outcomes of each pregnancy, 25% chance the child inherits two 3:03 normal alleles, 3:05 so they're unaffected and they're not even a carrier. 3:08 50% chance the child will inherit one normal allele and one mutated allele, and 3:13 so they 3:13 will become a carrier, but usually unaffected, no symptoms. 3:18 And then there's a 25% chance the child will inherit two mutated alleles and 3:23 develop PKU. 3:25 They will develop phenylketonuria. 3:30 Another example is if one parent was a carrier and one was actually affected 3:35 with PKU. 3:36 So here we have a PKU parent, MM, and the other parent, a carrier, so NM. 3:44 So the possible outcomes of each pregnancy, if we do the planet square, 50% 3:49 chance the 3:49 child will inherit two mutated alleles and so they will develop PKU. 3:55 But there's also a 50% chance the child will inherit one normal allele, but 3:59 always 4:00 one mutated, and so they will become a carrier and not show symptoms. 4:06 The third example is if one parent was a carrier and the other parent was a non 4:10 -carrier, so 4:11 they don't have any mutation in their Crohn's Zone 12. 4:15 So therefore, one parent will be NM for being a carrier and the other one will 4:21 be NM for 4:22 normal normal. 4:24 So the possible outcomes of each pregnancy, 50% chance the child will inherit 4:29 one normal 4:29 allele and one mutated allele and so they will become a carrier. 4:34 But again, typically no symptoms and 50% chance the child will inherit two 4:39 normal alleles from 4:40 each parent and so they will become unaffected, a non-carrier. 4:46 Last example is if two parents, both of whom have PKU, so both have MM, 4:51 mutation mutation 4:53 in both alleles, well the only outcome of each pregnancy is that there's 100% 4:58 chance 4:59 the child will inherit two mutated alleles and so the child will develop PKU. 5:04 I hope this planet square diagrams make sense in terms of how a phenylketonuria 5:12 or PKU is 5:14 inherited by either not parents with no mutations at all, a carrier or who have 5:21 PKU. 5:22 Now the risk of developing PKU is actually the same for both females and men. 5:30 So clinical features of one who develops PKU, well infants with PKU often show 5:35 no manifestations 5:37 of disease at birth and with early screening and management individuals may 5:41 never show 5:41 symptoms of the disease, however newborns that are not diagnosed early may be 5:46 weak and 5:47 have poor feeding. 5:48 Other symptoms, vomiting, irritability, rash are some other features. 5:54 Mental delay may present at several months, intellectual disability can occur 5:58 and is a 5:59 result of increased phenylalanine levels in the brain which causes destruction 6:04 of the 6:04 myelin of nerve fibers. 6:07 Phenylalanine levels can also cause depression due to reduction in dopamine 6:11 level and serotonin 6:12 level. 6:18 Other symptoms if people become untreated long term includes development of 6:23 light eye, skin 6:24 and hair color and this is due to the alteration in melanin production, a musty 6:29 odor and that's 6:31 caused by the phenyl acidic acid in the urine and the sweat and of course 6:36 neurological symptoms 6:38 because increased level of phenylalanine which is not converted to tyrosine 6:43 buildup and can 6:44 cause neurological problems including seizures, abnormal muscle movements, 6:49 tight muscles, 6:50 increased reflexes, involuntary movements or tremors. 6:54 Complications of PKU if again untreated irreversible brain damage and 7:00 intellectual disability, 7:02 neurological problems that I've outlined previously, behavioral problems and 7:07 psychological issues 7:08 such as depression due to low dopamine. 7:15 So the diagnosis and investigation of PKU can be done with routine newborn 7:20 screening which 7:22 measures an elevated level of phenylalanine on a blood spot. 7:28 Alternatively PKU can be diagnosed with molecular genetic testing that shows 7:32 two disease-causing 7:33 variants of the PAH gene. 7:37 Remember you need two mutations in both the chromosomes in order to develop 7:42 phenylketonuria 7:44 PKU. 7:50 The management of PKU involves maintaining plasma phenylalanine levels within a 7:55 safe range to 7:56 prevent the buildup and to prevent intellectual disability as well as 8:01 neurological, behavioral 8:03 and skin issues, this can actually generally be done through lifelong dietary 8:09 management. 8:10 As phenylalanine is found in protein rich foods, generally high protein foods 8:14 such as 8:15 meat, milk, fish and cheese are not allowed and naturally low protein foods, 8:19 fruits, vegetables 8:20 and some cereals can be allowed in limited quantities. 8:24 Phenylalanine free amino acid supplements enriched with micronutrients are 8:28 essential 8:29 to meet protein requirements. 8:32 The monitoring of phenylalanine levels in the blood is essential. 8:41 There are other potential treatments available such as seperopteran or QVAN. 8:46 This works as a cofactor for the phenylalanine hydroxylase enzyme. 8:55 It stimulates essentially the residual phenylalanine hydroxylase enzyme to 9:01 metabolize phenylalanine 9:03 into tyrosine. 9:04 However, only a few people with PKU are responsive to this treatment. 9:09 Another treatment is pigvallease PQPZ or palensic. 9:16 It's an injectable enzyme therapy. 9:18 It substitutes for the deficient phenylalanine hydroxylase enzyme in 9:24 individuals with PKU, 9:25 facilitating again the breakdown of phenylalanine into less harmful substances, 9:30 thereby reducing 9:31 its accumulation in the body. 9:38 If adequate treatment is started within the first days of life, severe 9:43 manifestations 9:44 of the disease are prevented, however mental health issues and mild cognitive 9:47 deficits 9:48 can still occur even with good control. 9:51 If treatment is delayed for two to three years, it may only be effective for 9:54 controlling the 9:55 extreme hyperactivity and intractable seizures. 9:59 If children are born to mothers that have poorly controlled PKU, they are at a 10:03 high risk 10:04 of developmental deficits and microcephaly. 10:08 So PKU does not shorten life expectancy with or without treatment importantly. 10:18 So in summary, today we talked about phenylketone urea, a genetic disorder that 10:23 is caused 10:24 by an inborn era metabolism, which affects the phenylalanine hydroxylase gene, 10:29 resulting 10:30 in absence or deficiency of the phenylalanine hydroxylase enzyme, an enzyme 10:35 which is responsible 10:37 again for the breakdown of the amino acid phenylalanine. 10:41 We discussed the genetics, clinical features of PKU as well as the diagnosis, 10:45 management 10:46 and prognosis, thank you.