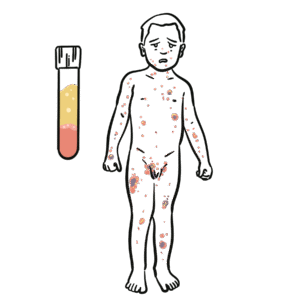
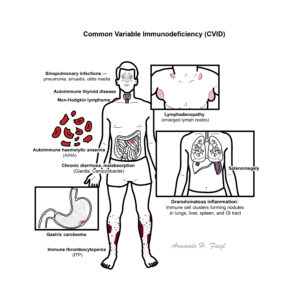
Common Variable Immunodeficiency (CVID) is the most frequent clinically significant primary antibody deficiency, characterised by hypogammaglobulinaemia and impaired antibody responses. It typically presents with recurrent bacterial infections, autoimmunity, granulomatous disease, and an increased risk of malignancy. CVID affects both sexes equally, with a prevalence of ~1 in 25,000–50,000 individuals. Diagnosis is often delayed, with onset usually in childhood or early adulthood. Despite “common” in its name, it remains rare, though more frequently encountered than severe combined immunodeficiency (SCID) or chronic granulomatous disease (CGD).
Definition
Hypogammaglobulinaemia: Abnormally low levels of serum immunoglobulins (IgG ± IgA/IgM). Primary immunodeficiency: Genetic or idiopathic immune system defect leading to recurrent or unusual infections. Granulomatous disease: Localised aggregation of immune cells (macrophages, T-cells) in response to persistent antigenic stimulation. Autoimmunity: Failure of self-tolerance leading to immune attack on host tissues (e.g., autoimmune cytopenias, thyroid disease).
CVID
C – Chronic/recurrent infections
V – Variable presentation (autoimmunity, granulomas, GI disease)
I – Immunoglobulin low (IgG ± IgA/IgM)
D – Defective antibody response
Anatomy & Physiology
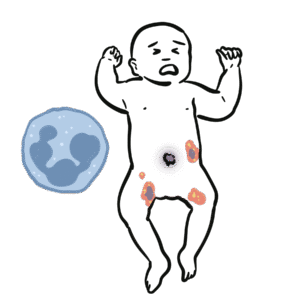
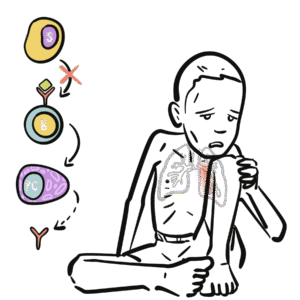
Normal physiology: B-cells mature in bone marrow, then differentiate into plasma cells that produce immunoglobulins (IgG, IgA, IgM, IgE).
Immunoglobulins provide:
IgG → systemic protection, memory response
IgA → mucosal immunity (respiratory, gut)
IgM → first-line antibody in acute infections
In CVID → failure of B-cell differentiation and impaired class switching → reduced immunoglobulin levels and poor vaccine responses.
Unlike X-linked agammaglobulinaemia (absent B-cells), in CVID B-cells are present but dysfunctional.
Aetiology
Exact cause unknown; multifactorial.
Genetic associations: mutations in ICOS, TACI (TNFRSF13B), BAFF-R, CD19, CD20, CD21, CD81 in some cases.
Risk Factors
Family history of primary immunodeficiency.
Certain HLA haplotypes associated with autoimmunity in CVID.
Higher risk in Caucasian populations.
Pathophysiology
Genetic/idiopathic defect in B-cell signalling or T-cell–B-cell interaction.
Failure of terminal B-cell differentiation → impaired plasma cell formation.
Hypogammaglobulinaemia (↓ IgG, ± IgA/IgM).
Poor vaccine response and inability to mount protective antibodies. Consequences: recurrent infections, autoimmunity, granulomatous inflammation, increased malignancy risk.
CVID = “B-cells present, but antibodies absent or dysfunctional.”
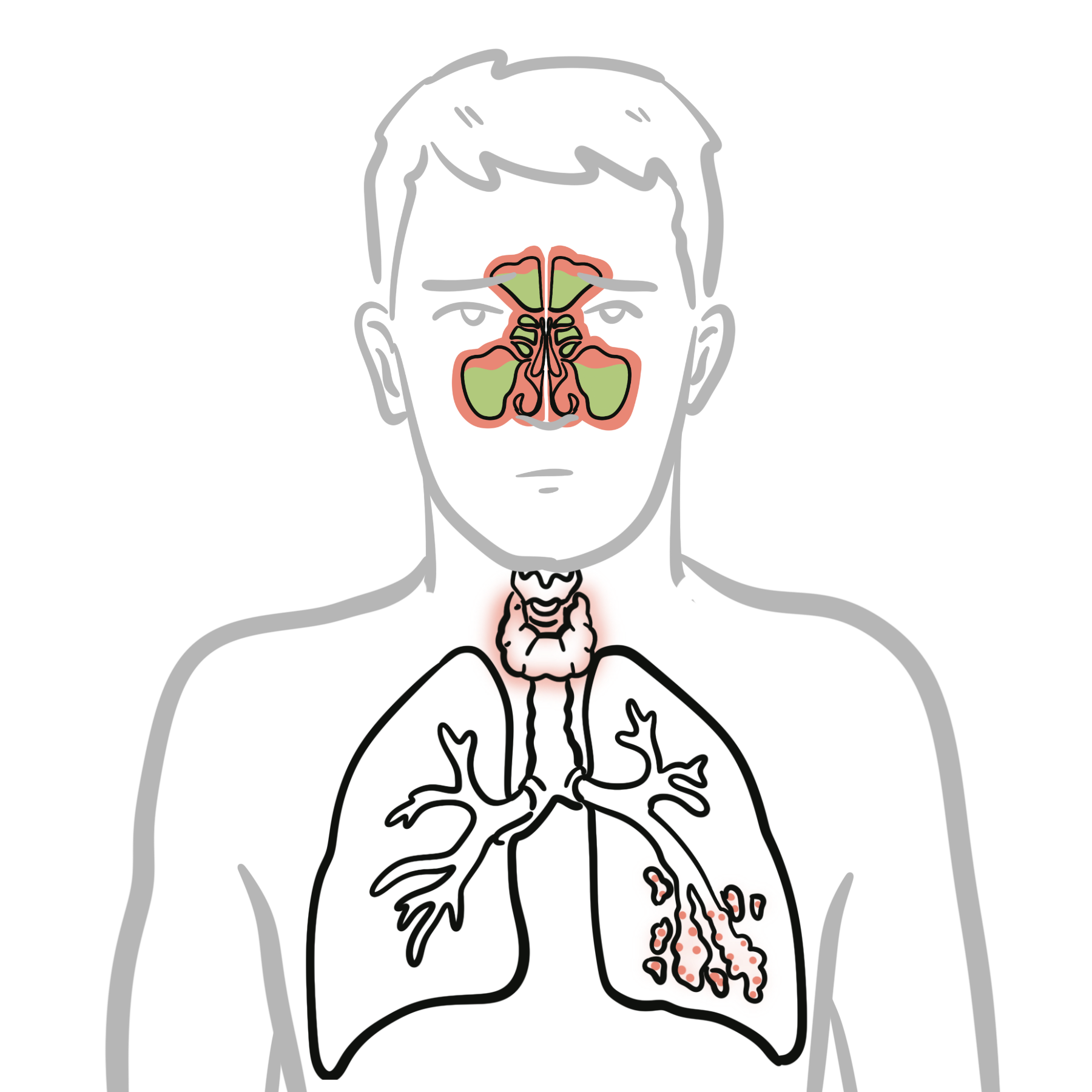
Recurrent pneumonia with established bronchiectasis.
Mortality often due to chronic lung disease or malignancy rather than acute infection.
References
Bonilla FA, Barlan I, Chapel H, et al. International Consensus Document (ICON): Common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2016;4(1):38–59.
Gathmann B, Mahlaoui N, Gérard L, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014;134(1):116–26.
Cunningham-Rundles C. How I treat common variable immune deficiency. Blood. 2010;116(1):7–15.
Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–86.
Yazdani R, Abolhassani H, Aghamohammadi A. Common variable immunodeficiency: Epidemiology, pathogenesis, clinical manifestations, diagnosis, classification, and management. J Investig Allergol Clin Immunol. 2020;30(1):14–34.

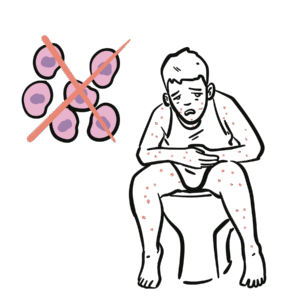








Discussion