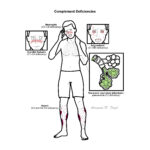
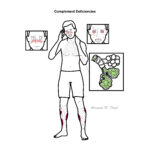
Complement deficiencies are rare primary immunodeficiencies resulting from inherited absence or dysfunction of complement proteins, regulators, or receptors. They predispose to recurrent bacterial infections, autoimmune disease (particularly systemic lupus erythematosus–like syndromes), and in some cases angioedema. Complement proteins play a central role in innate immunity by opsonisation, chemotaxis, and direct lysis of pathogens via the membrane attack complex (MAC). Overall prevalence is low but varies depending on the specific pathway; deficiencies of terminal components (C5–C9) are strongly associated with recurrent Neisseria infections.
Definition
Complement system: A group of plasma proteins that enhance immune response via opsonisation, inflammation, and cell lysis. Classical pathway: Activated by antigen–antibody complexes. Alternative pathway: Antibody-independent activation, triggered by pathogen surfaces. Membrane attack complex (MAC): Terminal complex (C5b–C9) that creates pores in bacterial membranes, leading to lysis.
Anatomy & Physiology
Three activation pathways:
Classical → triggered by IgG/IgM immune complexes.
Lectin → triggered by mannose-binding lectin binding microbial carbohydrates.
Alternative → continuous low-grade activation on microbial surfaces.
All pathways converge at C3 convertase → C3b opsonisation, C5 convertase activation → C5b-9 MAC formation.
Complement also enhances phagocytosis and recruits inflammatory cells (C3a, C5a = anaphylatoxins).
C1-INH concentrate or bradykinin inhibitors for hereditary angioedema.
Genetic counselling.
Vaccination against Neisseria meningitidis is essential in terminal pathway deficiencies.
Complications & Prognosis
Early pathway deficiency → recurrent infections + high risk SLE/autoimmunity.
C3 deficiency → poor prognosis without prophylaxis due to severe recurrent sepsis.
Terminal pathway deficiency → recurrent but survivable Neisseria infections if recognised and vaccinated.
Hereditary angioedema → risk of life-threatening airway obstruction.
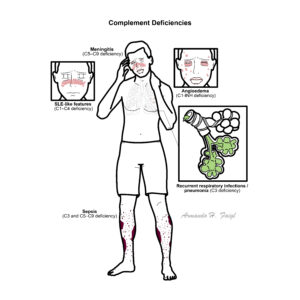
Complement Deficiency Patterns
Deficiency
Typical Presentation
Key Pathogens
Associated Conditions
C1, C2, C4
Recurrent infections + SLE-like disease
Pyogenic bacteria
SLE, autoimmune disease
C3
Severe recurrent infections
S. pneumoniae, H. influenzae
Sepsis, meningitis
C5–C9
Recurrent meningitis
Neisseria meningitidis
–
C1 inhibitor
Angioedema (non-pruritic, airway risk)
–
Hereditary angioedema
References
Walport MJ. Complement. N Engl J Med. 2001;344(14):1058–66.
Skattum L, van Deuren M, van der Poll T, Truedsson L. Complement deficiency states and associated infections. Mol Immunol. 2011;48(14):1643–55.
Grumach AS, Kirschfink M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Mol Immunol. 2014;61(2):110–7.













Discussion