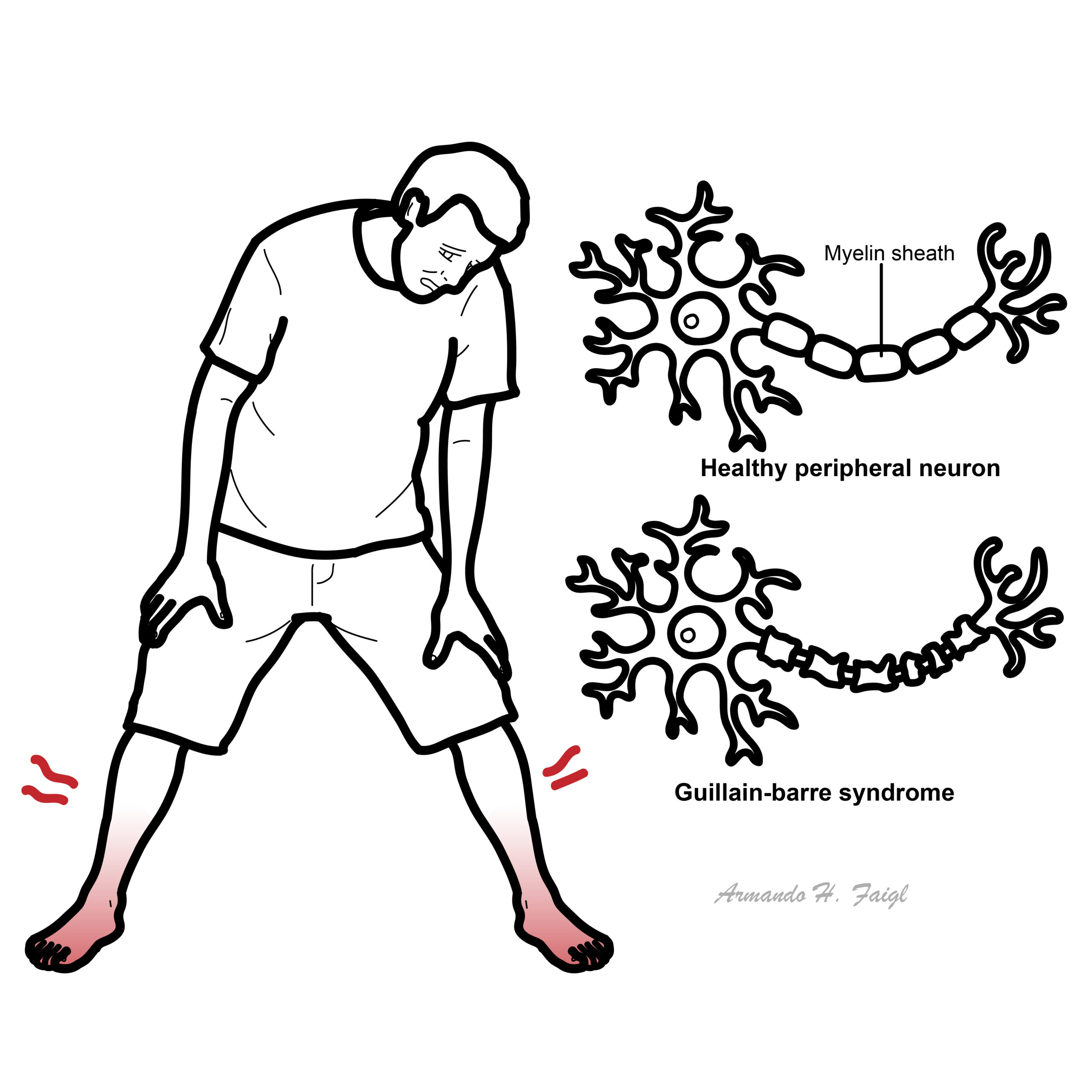
Guillain-Barré Syndrome (GBS) is an acute, immune-mediated polyneuropathy typically triggered by a preceding gastrointestinal or respiratory infection. It is characterised by an autoimmune attack on the peripheral nerves, leading to mainly demyelination but also axonal degeneration, which impairs nerve conduction and causes muscle weakness and paralysis. GBS affects about 1–2 per 100,000 people annually. People of all ages can be affected, but it is more common in adults and in males.
Definition
Acute inflammatory demyelinating polyneuropathy (AIDP): demyelinating form, common in Western countries, good prognosis. Acute Motor Axonal Neuropathy (AMAN): axonal form, more severe, often associated with C. jejuni, slower recovery. Miller Fisher syndrome: ophthalmoplegia, ataxia, areflexia Albuminocytologic dissociation: presence of increased CSF protein without a corresponding increase in CSF white blood cells.
Aetiology and Risk Factors
GBS is a post-infectious acute auto-immune mediated neuropathy that is typically preceded by a gastrointestinal or respiratory tract infection with the most common pathogens including:
Campylobacter Jejuni (seen in 20-35% of cases)
Cytomegalovirus (CMV)
Epstein-Barr virus
Mycoplasma Pneumoniae
Antecedent infections (especially Campylobacter jejuni) are common – ask about diarrhoea or respiratory illness.
Pathophysiology
GBS is often preceded by a gastrointestinal or URT infection which triggers an autoimmune response against nerve sheath and axons. The main mechanism is thought to be through molecular mimicry, whereby the most common organism C. jejuni has surface antigens that resemble gangliosides on peripheral nerves. As a result, the body creates ganglioside autoantibodies, of which exist multiple variants, that attack components of the myelin sheath or axons of peripheral nerves.
In the most common variant, acute inflammatory demyelinating neuropathy (AIDP), immune cell infiltration and macrophage-mediated stripping of the myelin sheath causes segmental demyelination, resulting in poor nerve conduction (conduction block) and subsequent muscle weakness and flaccid paralysis. In this variant however there is potential for full recovery. In the acute motor axonal neuropathy (AMAN) variant (more common in C. Jejuni infections), the immune attack is centred at the nodes of Ranvier, resulting in direct axonal degeneration. AMAN has a more severe and prolonged recovery time.1, 2, 3, 4
Clinical Manifestations
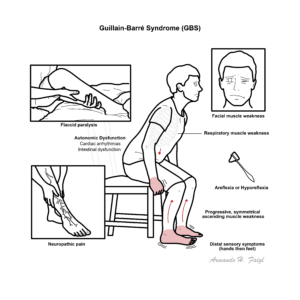
GBS typically presents as progressive, symmetrical, ascending muscle weakness that reaches nadir by about the 4th week of infection. Patients commonly experience flaccid paralysis, varying ranges of areflexia or hyporeflexia and a distal-proximal progression of weakness. It often causes sensory changes in the hands followed by the feet. Cranial nerve involvement involves the facial and bulbar muscles leading to dysphagia and facial muscle weakness. Patients are also likely to experience neuropathic pain.
Up to two thirds of patients experience autonomic involvement which can manifest In the form of cardiac arrhythmias, hyper/hypotension ileus and urinary incontinence. Respiratory muscle weakness is seen in 25% of cases and has resulted in some population groups requiring mechanical ventilation.1, 5, 6, 7
Miller Fisher syndrome is a rare variant of GBS characterised by a distinct triad of:
Ophthalmoplegia (paralysis of eye movements)
Ataxia (gait and limb incoordination)
Areflexia (absent reflexes)
This image series is only available to members
Miller Fisher syndrome triad: ophthalmoplegia, ataxia, areflexia. Strongly associated with anti-GQ1b antibodies.
GBS typically presents with progressive, symmetrical weakness that starts in the legs and ascends (ascending paralysis), areflexia or hyporeflexia is a hallmark early sign, but the disease can present atypically or as a clinical variant.
Diagnosis
GBS is considered to be a clinical diagnosis that can be made on bedside if suspicion is high. Further investigations can be done to rule out differentials and determine the variant of GBS, but no investigation is specific in the diagnosis.1, 7, 8
Cerebrospinal fluid testing: Raised CSF proteins but may be normal in the first week of infection. WCC is usually normal in most cases
Raised CSF proteins with normal WCC is termed albuminocytologic dissociation
Nerve conduction studies: Can help distinguish between demyelination and axonal disease
Findings may include: Conduction blocks, prolonged or absent “F” waves
Anti-ganglioside antibodies
Anti-GM1: worse prognosis
Anti-GD3
Anti-GQ1b: associated with Miller Fisher syndrome
Has a smaller diagnostic role but may have a prognostic role
Other blood tests to exclude conditions that mimic GBS such as electrolyte deficiencies, porphyria and IgA deficiency
Nerve conduction studies show demyelinating features (prolonged F-wave latency, conduction block) in most GBS subtypes.
20% of patients may have a long termdisability and lose the ability to walk without aids
GBS is typically a monophasic illness, though a subset of patients may experience deterioration after initial stabilisation or improvement with treatment — a pattern known as treatment-related fluctuation (TRF).
True relapses of GBS occur in approximately 2–5% of cases
Bulbar palsy (risk of aspiration, swallowing difficulties)
Autonomic dysfunction (cardiac arrhythmias, BP instability)
References
National Institute of Neurological Disorders and Stroke. Guillain-Barré Syndrome [Internet]. Bethesda (MD): National Center for Biotechnology Information; 2018 [cited 2025 Jun 29]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532254/
Yuki N, Hartung HP. Guillain-Barré syndrome. N Engl J Med. 2012;366(24):2294–2304. doi:10.1056/NEJMra1114525
Brown WF, Feasby TE. Conduction block and denervation in Guillain-Barré polyneuropathy. Brain. 1984;107(Pt 1):219–239. doi:10.1093/brain/107.1.219
Leonhard SE, Mandarakas MR, Gondim FAA, et al. Diagnosis and management of Guillain-Barré syndrome in ten steps. Nat Rev Neurol. 2019;15(11):671–683. doi:10.1038/s41582-019-0250-9















Discussion