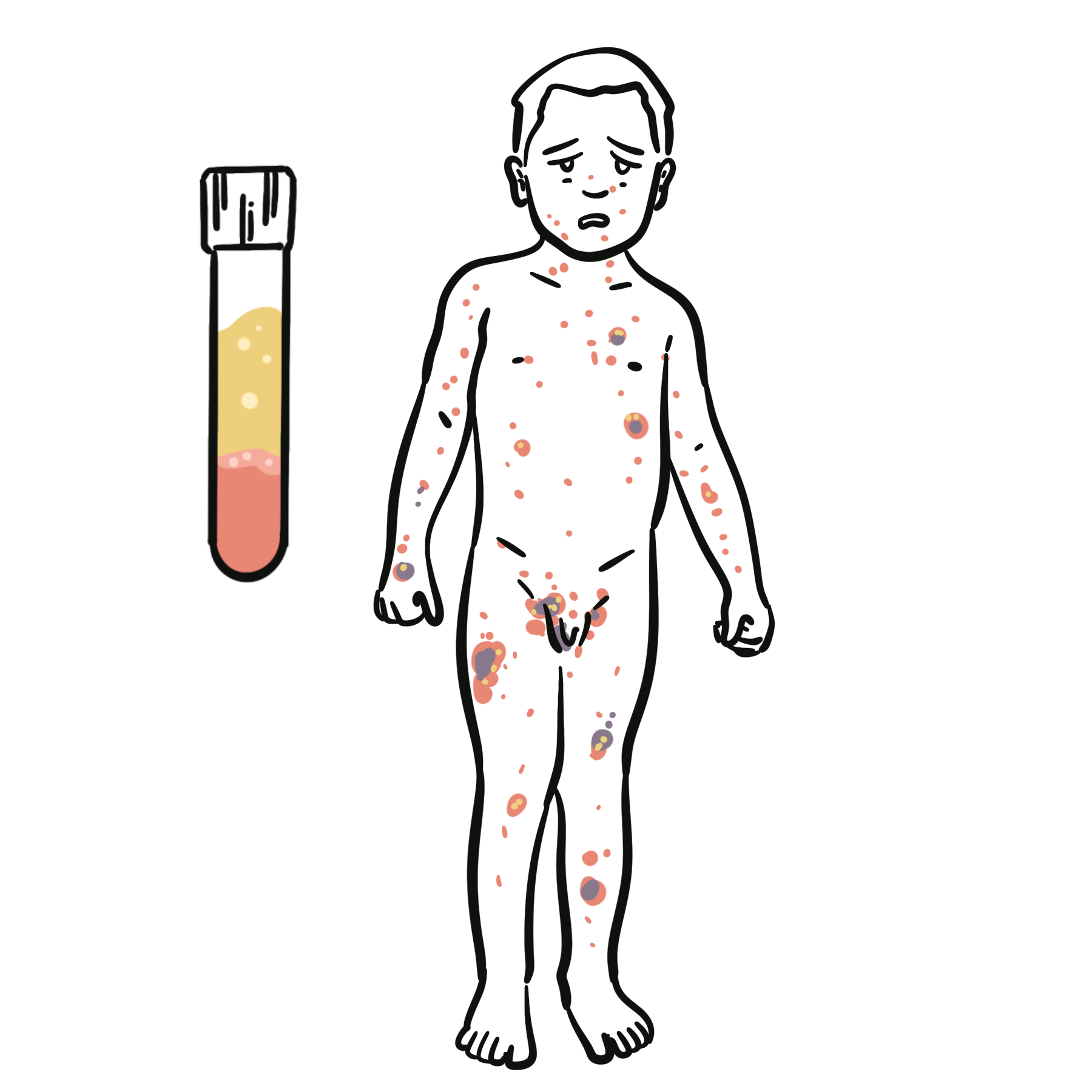
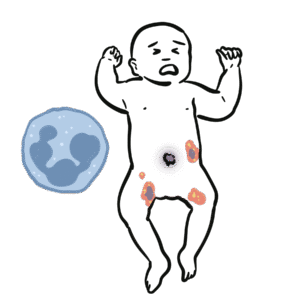
Selective IgA deficiency is the most common primary immunodeficiency, defined by very low or absent serum IgA levels with normal levels of other immunoglobulins (IgG, IgM). Many patients are asymptomatic, but some develop recurrent mucosal infections, allergic disease, or autoimmune disorders. Prevalence is estimated at 1:300 to 1:700 in Caucasian populations. Diagnosis is often incidental on routine blood tests, though symptomatic cases usually present in childhood or early adulthood.
Definition
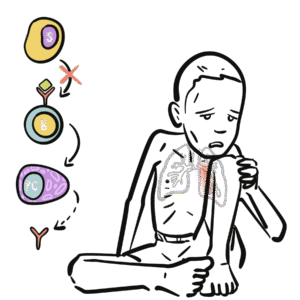
IgA: Immunoglobulin found in mucosal secretions (respiratory tract, gut, saliva, breast milk) – provides frontline defence. Primary immunodeficiency: Genetic/idiopathic defect of the immune system. Anti-IgA antibodies: Antibodies directed against IgA, can cause anaphylaxis during blood transfusion. Asymptomatic carrier: Patient with immunological abnormality but no clinical manifestations.
Anatomy & Physiology
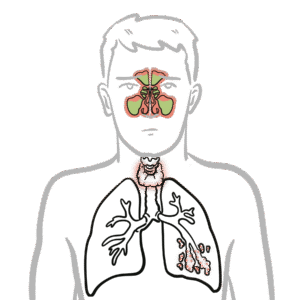
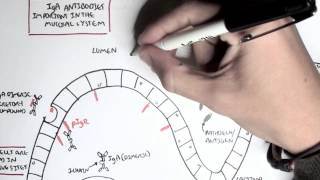
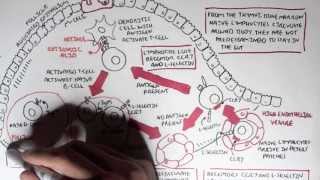
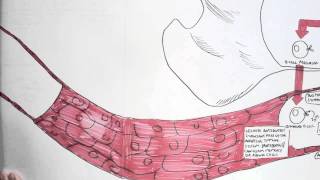
IgA physiology: Produced by plasma cells in mucosa-associated lymphoid tissue (MALT).
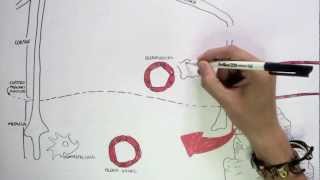
Secretory IgA (sIgA): Dimeric form secreted across mucosal epithelia; protects against pathogens by neutralisation and blocking adhesion.
IgA also regulates microbiota balance and mucosal tolerance.
In SIgAD → absent/low IgA → impaired mucosal immunity → recurrent respiratory/GI infections and increased autoimmunity risk.
IgA is the first line of defence at mucosal surfaces.
SIgAD often found incidentally, but autoimmune disease and transfusion reactions are the high-yield risks.
SIgAD vs CVID
Feature
SIgAD
CVID
IgA
↓/absent
↓ (± IgG/IgM)
IgG
Normal
↓
Vaccine response
Normal
Impaired
Age of onset
Often childhood/adolescence
Later (childhood–adulthood)
Autoimmunity
Common
Common
Progression
Can progress to CVID
Established immunodeficiency
References
Yel L. Selective IgA deficiency. J Clin Immunol. 2010;30(1):10–6.
Ludvigsson JF, Neovius M, Hammarström L. Association between IgA deficiency & other autoimmune conditions: a population-based matched cohort study. J Clin Immunol. 2014;34(4):444–51.
Wang N, Hammarström L. IgA deficiency: what is new? Curr Opin Allergy Clin Immunol. 2012;12(6):602–8.
Picard C, Al-Herz W, Bousfiha A, et al. Primary Immunodeficiency Diseases: IUIS Classification Update. J Clin Immunol. 2015;35(8):696–726.
Latiff AH, Kerr MA. The clinical significance of immunoglobulin A deficiency. Ann Clin Biochem. 2007;44(2):131–9.


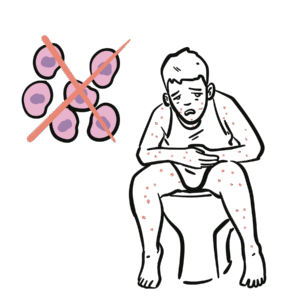









Discussion