

Behçet’s Disease

Overview
Behçet’s syndrome is a chronic, relapsing, multisystem inflammatory disorder and variable-vessel vasculitis characterized classically by recurrent oral ulcers, genital ulcers, ocular inflammation, and skin lesions. It can also involve the joints, vessels, nervous system, and gastrointestinal tract.
The disease is most prevalent along the historic “Silk Road” region, including Turkey, the Middle East, and East Asia, but occurs worldwide. Onset is usually in young adults, and major-organ disease tends to be more severe in men and in earlier-onset disease. Diagnostic delay is common because manifestations accumulate over time and there is no single confirmatory laboratory test.
Think of Behçet’s in recurrent oral ulcers plus any combination of genital ulcers, uveitis, skin lesions, thrombosis, or unexplained inflammatory neurologic disease.
Definition
Behçet’s syndrome: a multisystem inflammatory disorder/variable-vessel vasculitis affecting arteries and veins of any size.
Aphthous ulcer: painful, shallow mucosal ulcer with erythematous rim; recurrent oral aphthae are the hallmark mucosal lesion.
Pathergy: exaggerated skin hyperreactivity after minor trauma, classically producing a papule or pustule 24–48 hours after needle prick.
Uveitis: intraocular inflammation; in Behçet’s this may be anterior, posterior, or panuveitis, often with retinal vasculitis.
Aetiology & Risk Factors
Aetiology
- Exact cause is unknown. Current understanding supports environmental or microbial triggers acting in genetically predisposed individuals.
- HLA-B51 is the strongest genetic association, but it is supportive, not diagnostic.
- Disease pathogenesis involves abnormal innate and adaptive immune responses, particularly neutrophil hyperreactivity and Th1/Th17-skewed inflammation.
Risk factors / associations
- Geography and ancestry from high-prevalence regions increase pre-test probability.
- Male sex is associated with more severe ocular and vascular disease in many cohorts.
- Younger age at onset is associated with more severe major-organ involvement.
HLA-B51 helps support suspicion but cannot confirm or exclude Behçet’s.
Pathophysiology
- In a genetically susceptible host, environmental triggers activate innate immunity.
- Neutrophils and monocytes become hyperactive, with increased chemotaxis and pro-inflammatory cytokine production such as IL-1β, IL-6, and TNF-α.
- Dendritic-cell and cytokine signaling promotes Th1/Th17 responses, with increased IFN-γ, IL-17, and IL-23-related pathways.
- Endothelial inflammation and immune dysregulation then drive recurrent mucosal ulceration, neutrophilic skin lesions, ocular inflammation, and thrombosis-prone vascular disease.
- Repeated relapsing inflammation causes cumulative organ damage, especially visual loss and neurologic disability if major-organ disease is untreated.
Behçet thrombosis is usually driven by inflamed vessel wall rather than a conventional hypercoagulable state alone, which is why immunosuppression is central in vascular disease.
Clinical Manifestations
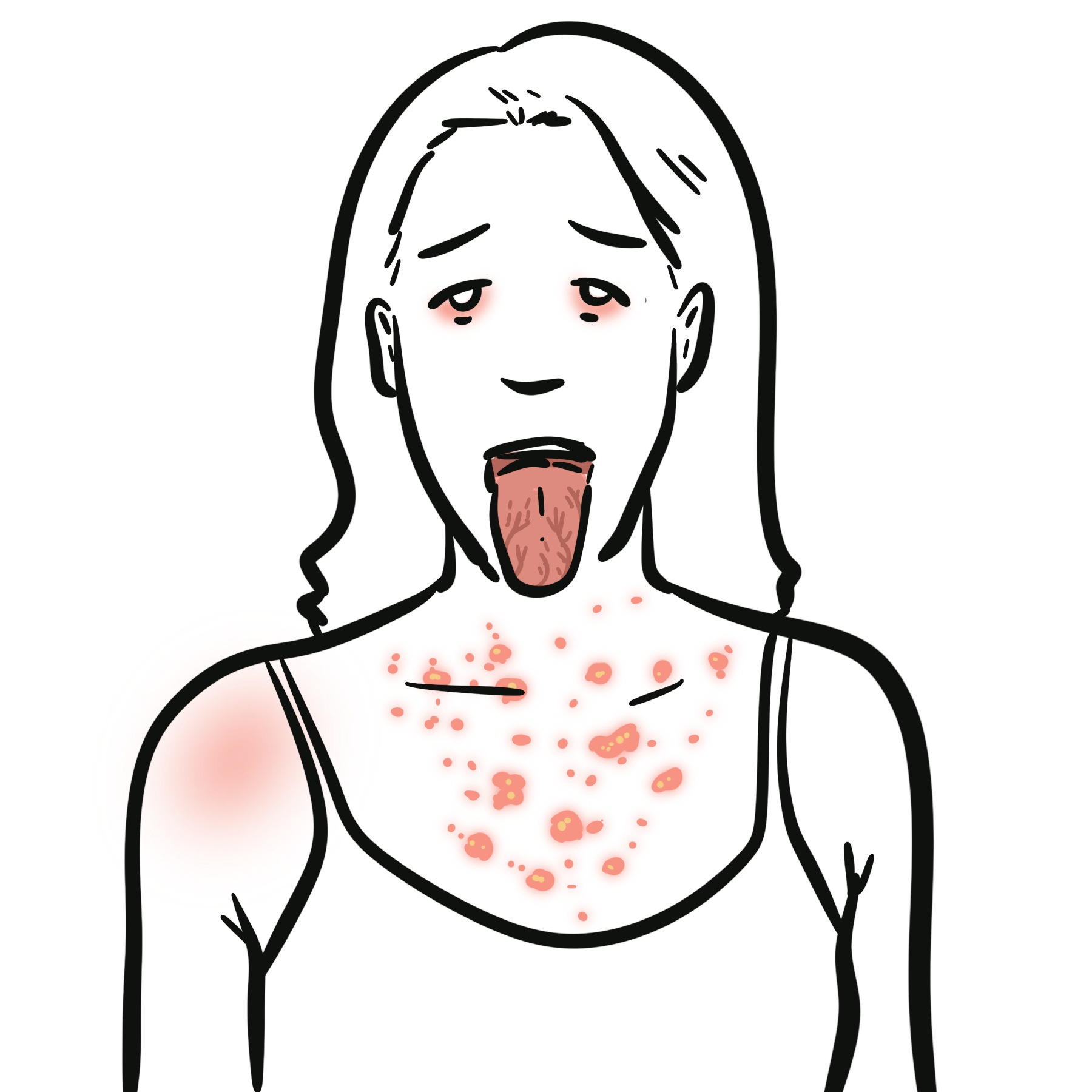
- Oral ulcers: recurrent, painful aphthous ulcers are the commonest and often earliest feature.
- Genital ulcers: painful recurrent ulcers, often healing with scarring.
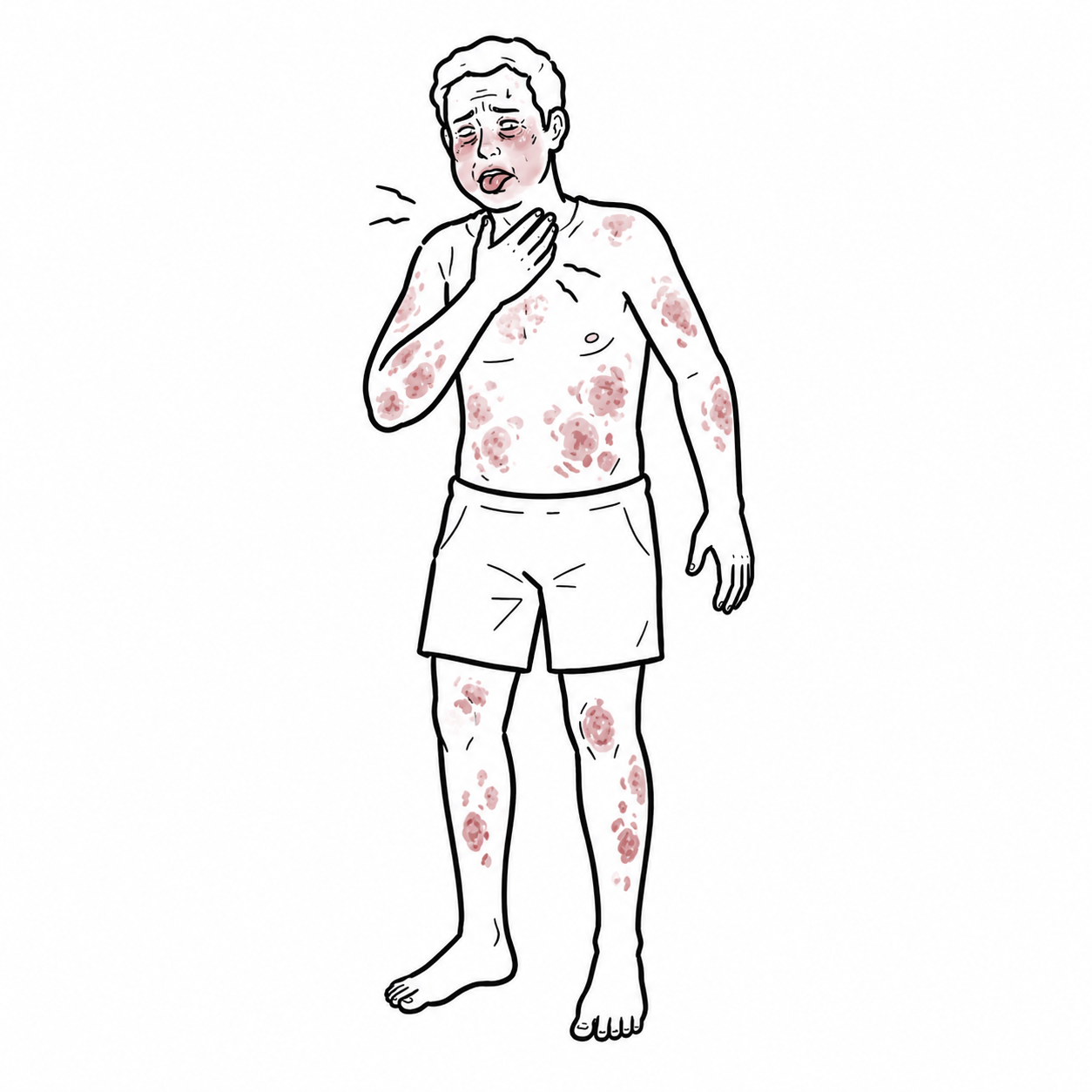
- Skin lesions: papulopustular lesions, pseudofolliculitis, acneiform lesions, erythema nodosum-like nodules.
- Eye disease: anterior or posterior uveitis, panuveitis, retinal vasculitis, floaters, blurred vision, painful red eye, reduced visual acuity.
- Musculoskeletal: non-erosive, intermittent inflammatory arthralgia or oligoarthritis, commonly knees/ankles/wrists.
- Vascular: superficial thrombophlebitis, deep-vein thrombosis, vena cava thrombosis, pulmonary artery aneurysm, arterial aneurysm or occlusion.
- Neurologic: headaches, brainstem syndrome, pyramidal signs, meningoencephalitis-like presentations, cerebral venous thrombosis.
- Gastrointestinal: abdominal pain, diarrhea, bleeding, deep round/oval ulcers especially in ileocecal region in intestinal Behçet’s.
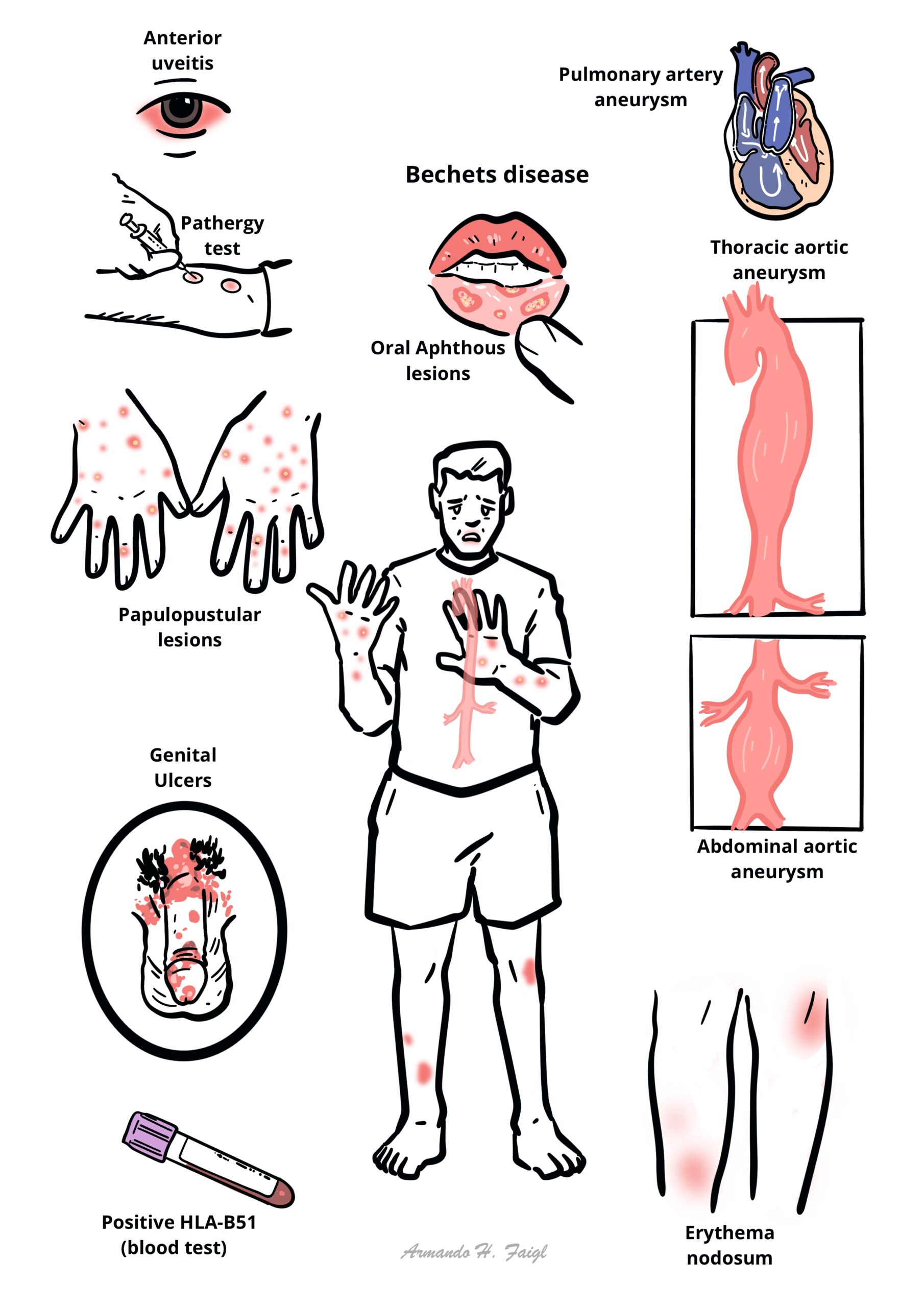
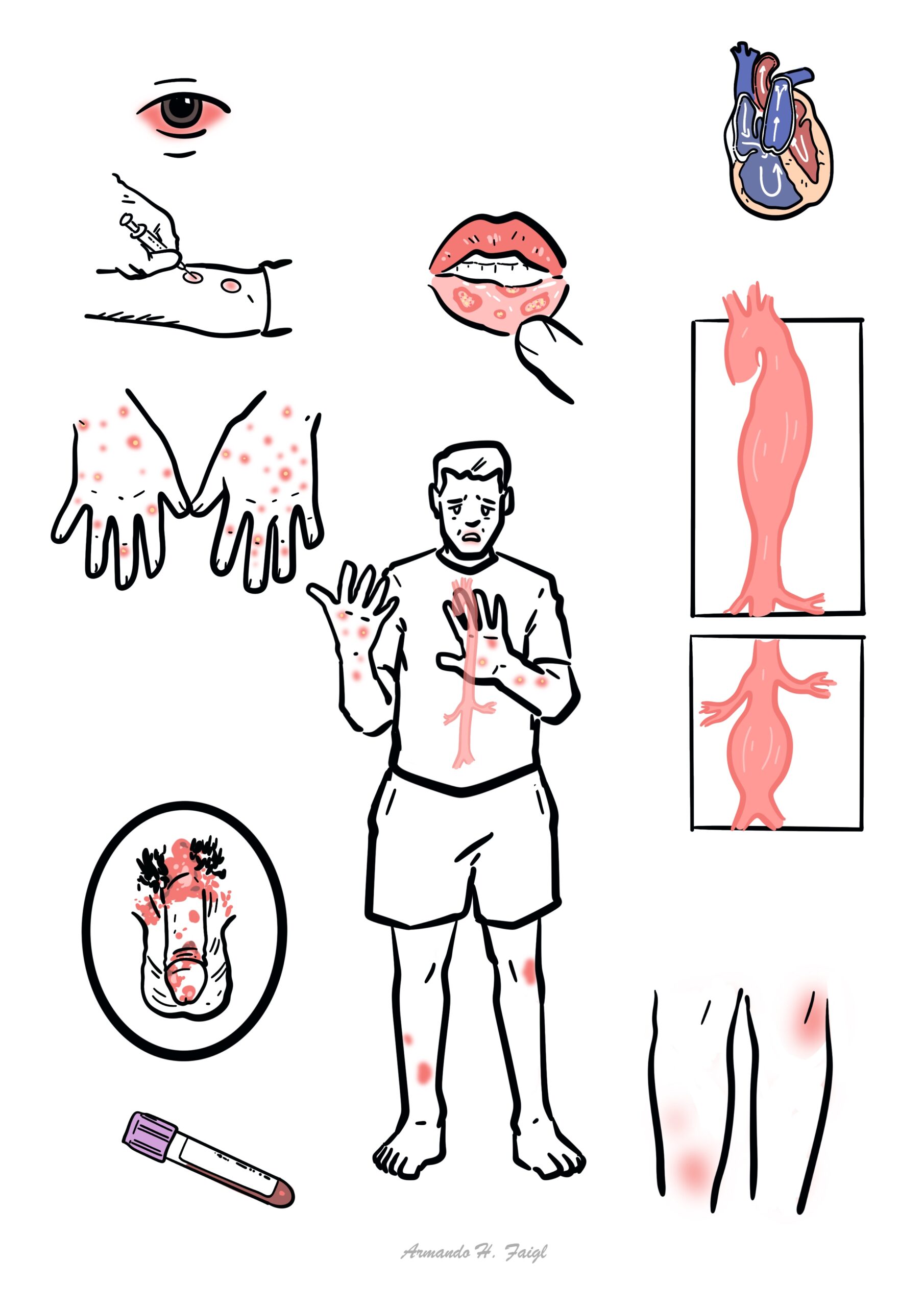
Classic triad: recurrent oral ulcers + genital ulcers + uveitis.
Important examination findings
- Active oral aphthae or scars from prior genital ulcers.
- Erythema nodosum-like lesions or papulopustules.
- Reduced visual acuity, hypopyon or signs of uveitis/retinal vasculitis on urgent ophthalmic examination.
- Signs of thrombosis, focal neurologic deficit, or GI bleeding as red flags for major-organ disease.
Cyclosporine seems to be associated with an increased risk of developing neurological parenchymal disease although the reason is unknown. Cyclosporine should be discontinued or avoided in patients with pNBD.
Behçet’s ulcers are typically painful and recurrent.
Eye symptoms in Behçet’s are an emergency because delay can cause irreversible visual loss.
Investigations
Behçet’s syndrome is based on the patient’s clinical presentation and imaging findings. Bechets is associated with HLAB51, but this is not part of the classification criteria. Whilst this is not used as a diagnostic tool, it is useful for supporting diagnosis.
The two most commonly used classifications for Behçet’s syndrome are the International Study Group for Behçet’s Disease (ISG) classification and the International Criteria for Behçet’s Disease (ICBD). A pathergy test is performed with at least three skin punctures, and a positive reaction is indicated by a papular reaction that is at least 2 mm in diameter and surrounded by erythema or the development of a pustule reaction within 24 to 48 hours.
| Table 1. Classification Criteria for Behçet’s Disease | ||
| ISG 1990 | ICBD 2014 | |
| Criteria | – Recurrent oral ulceration- Ocular lesions- Skin lesions- Positive pathergy test | Recurrent oral ulceration (2 points)Genital ulceration (2 points)Ocular lesions (2 points)Skin lesions (1 point)Vascular manifestations (1 point)Neurologic manifestations (1 point)Positive pathergy test (1 point) |
| Criteria or score needed for classification | Recurrent oral ulceration plus 2 other criteria | ≥4 points |
| Table 2 Organ involvement in Bechet’s and management | |
| Complication | Management |
| Skin | Colchicine (first line)Topical glucocorticoidsAntiinflammatory mouthwashesApremilast (recurrent mouth ulcers) |
| Ocular | Glucocorticoids TNFaIInterferon alpha Azathioprine |
| Vascular | Include vasculitis, thrombosis and aneursumGlucocorticoids to reduce inflammation with commencement of either: azathioprine or cyclophosphamideTNFaI for refractory disease Anticoagulation for thrombosis |
| Neurological | Glucocorticoids with slow taperingAzathioprineCyclosporine should be avoidedTNFaI for refractory diseaseAnticoagulation for thrombosis |
| Gastrointestinal | EndoscopyNSAID ulcers, inflammatory bowel disease and infections such as tuberculosis should be ruled out |
| Other Manifestations | Arthritis: Colchicine |
Schnitzler’s syndrome is a rare, sporadic, acquired autoinflammatory disorder that typically emerges in mid-adulthood. It is characterized by the pentad:
- Recurrent fever
- Chronic non-pruritic urticaria
- Arthritis
- Lymphadenopathy
- IgM gammopathy
Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) syndrome is an autoinflammatory disorder characterized by suppurative hidradenitis and acne starting in adolescence, with pyoderma gangrenosum (PG) developing later. Unlike PAPA syndrome, PASH lacks pyogenic arthritis. It is caused by mutations in PSTPIP1, via increased binding affinity to pyrin, that are responsible for the activation of an inflammasome.
VEXAS and SAPHO are discussed in a separate chapters but are classified as multifactorial autoinflammatory syndromes.
Treatment
- Treatment is organ-based and severity-based. Major-organ disease needs rapid specialist-directed immunosuppression.
Mucocutaneous / mild joint disease
- Topical corticosteroids for oral or genital ulcers.
- Colchicine is a key first-line drug for recurrent mucocutaneous lesions and arthritis, especially erythema nodosum-like lesions.
- Short systemic corticosteroids may be used for flares.
- Azathioprine, apremilast, TNF inhibitors, or interferon-alpha may be considered for refractory disease.
Ocular disease
- Posterior uveitis, retinal vasculitis, and panuveitis require urgent systemic therapy.
- EULAR-backed approaches favor azathioprine, cyclosporine-A, interferon-alpha, and monoclonal TNF inhibitors such as infliximab or adalimumab for sight-threatening disease.
Vascular disease
- Immunosuppression is central. Severe arterial disease and extensive venous disease may require glucocorticoids with agents such as cyclophosphamide or TNF inhibitors depending on phenotype.
- Anticoagulation in venous thrombosis is controversial and individualized; it is not a substitute for controlling vascular inflammation.
Neuro-Behçet’s
- High-dose glucocorticoids are typically used acutely. Azathioprine, cyclophosphamide, methotrexate, or TNF inhibitors may be used depending on severity and pattern. Cyclosporine is generally avoided in parenchymal neuro-Behçet’s because of concern for neurotoxicity.
Intestinal disease
- 5-ASA, corticosteroids, thiopurines, and biologics may be used depending on severity; management often overlaps with inflammatory bowel disease pathways but remains phenotype-specific.
Remember - Colchicine is high-yield for mucocutaneous Behçet’s.
- Sight-threatening eye disease needs urgent systemic therapy, not topical treatment alone.
Complications & prognosis
- Visual loss or blindness from recurrent posterior uveitis/retinal vasculitis.
- Venous thrombosis, pulmonary artery aneurysm, and major arterial complications.
- Neurologic disability from parenchymal neuro-Behçet’s or cerebral venous thrombosis.
- GI bleeding or perforation in intestinal Behçet’s.
- Chronic pain, fatigue, scarring, and psychosocial burden.
- Disease is relapsing-remitting; many patients do well with treatment, but major-organ disease drives morbidity. Severity often lessens over time in some patients, though irreversible damage can occur if organ-threatening disease is missed or undertreated.



Members only discussions coming soon…