

VEXAS Syndrome
Overview
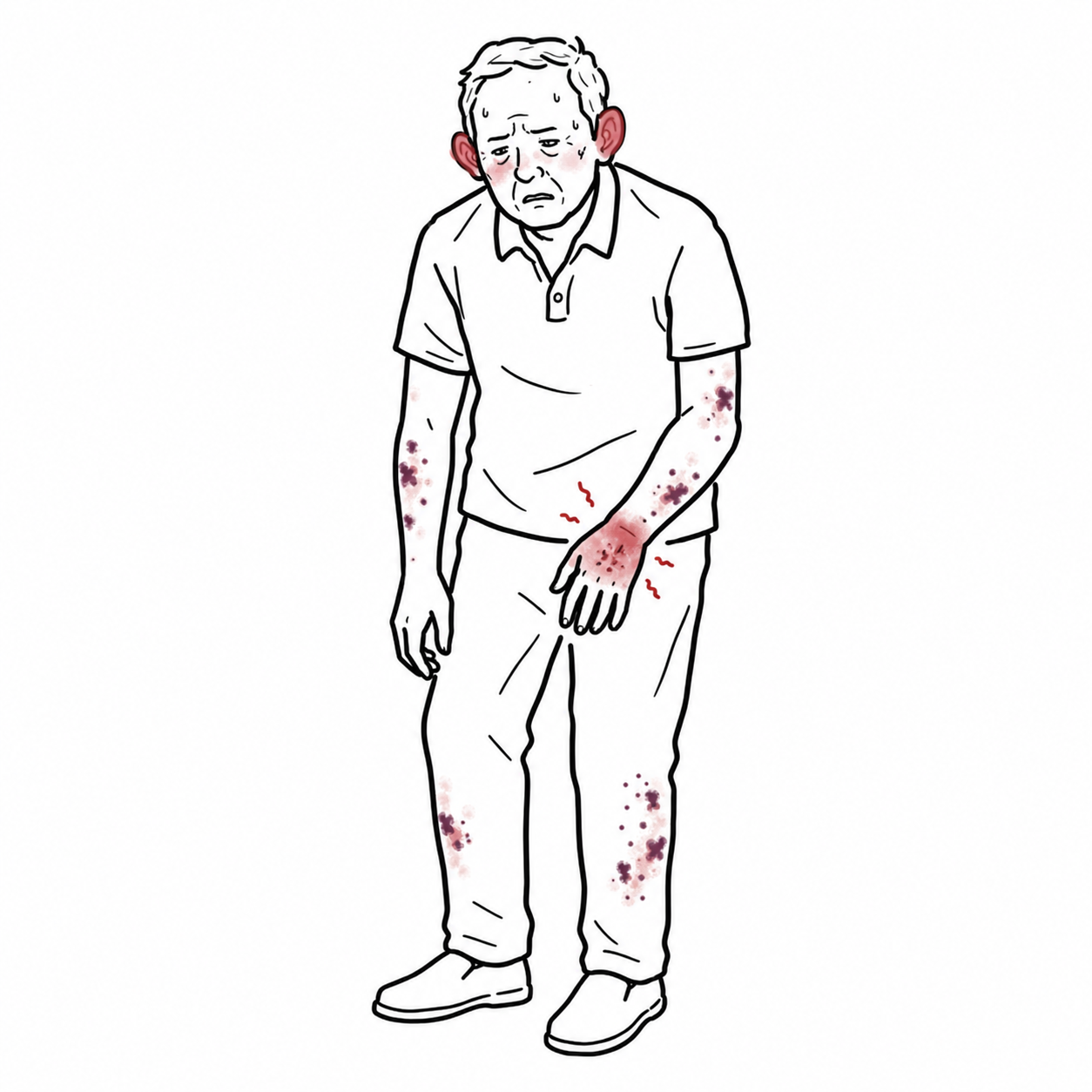
VEXAS syndrome is a late-onset, adult autoinflammatory disease caused by acquired somatic mutations in the UBA1 gene, leading to dysregulated innate immunity, systemic inflammation, and overlapping haematologic and rheumatologic manifestations.
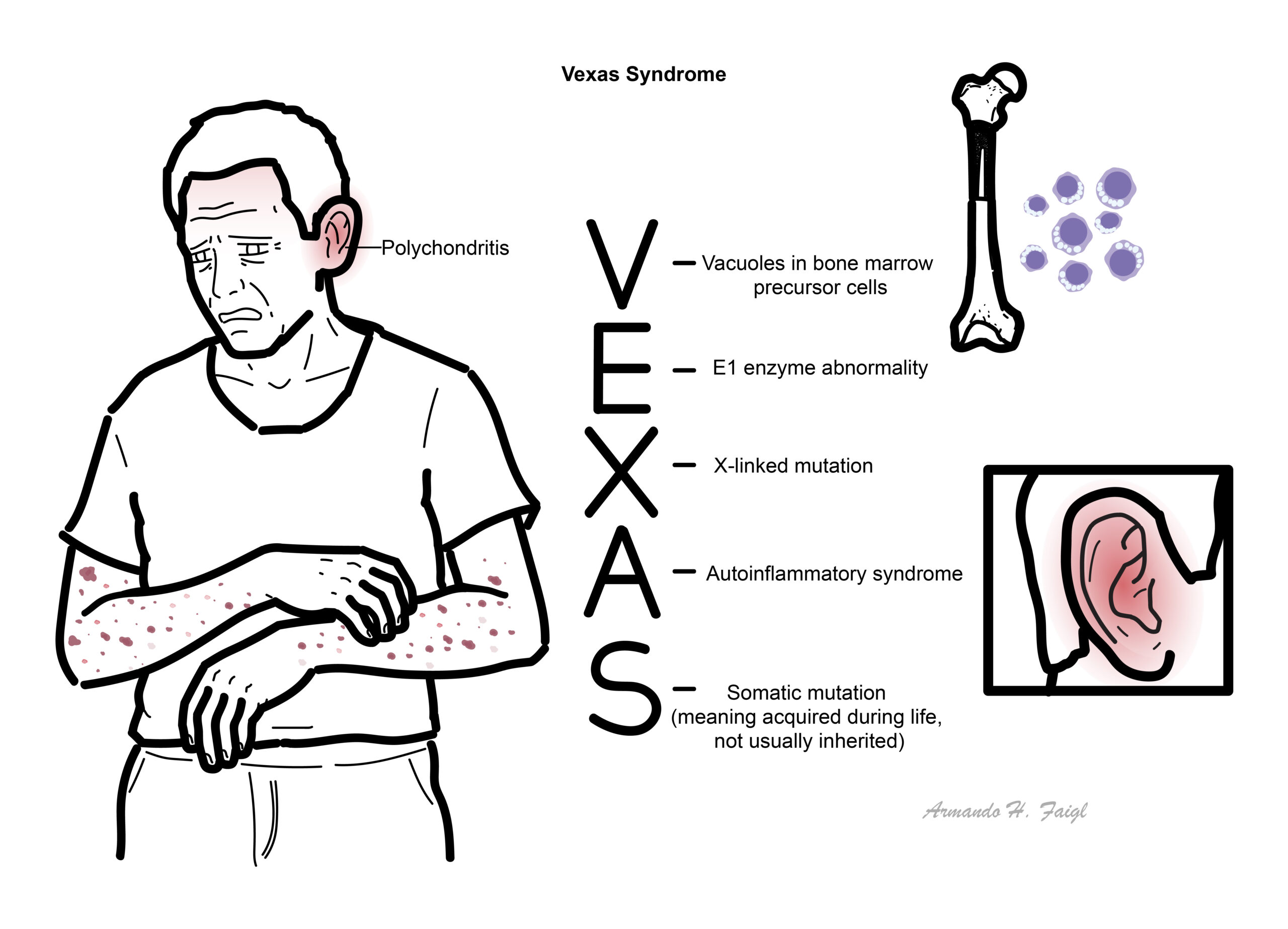
VEXAS stands for Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic.
It predominantly affects older males due to its X-linked nature and typically presents with refractory inflammation, cytopenias, and bone marrow abnormalities. The condition is increasingly recognised since its description in 2020, with many patients previously misclassified under other inflammatory or haematologic diseases.
Definition
UBA1 gene: Encodes ubiquitin-activating enzyme E1, essential for protein degradation and cellular regulation.
Somatic mutation: Acquired mutation (not inherited) occurring in haematopoietic stem cells.
Autoinflammatory disease: Disorder driven by innate immune dysregulation without autoantibodies.
Myelodysplastic syndrome (MDS): Clonal bone marrow disorder with ineffective haematopoiesis and cytopenias.
VEXAS is not inherited—it is acquired later in life and is associated with macrocytic anaemia and bone marrow vacuoles.
Anatomy & Physiology
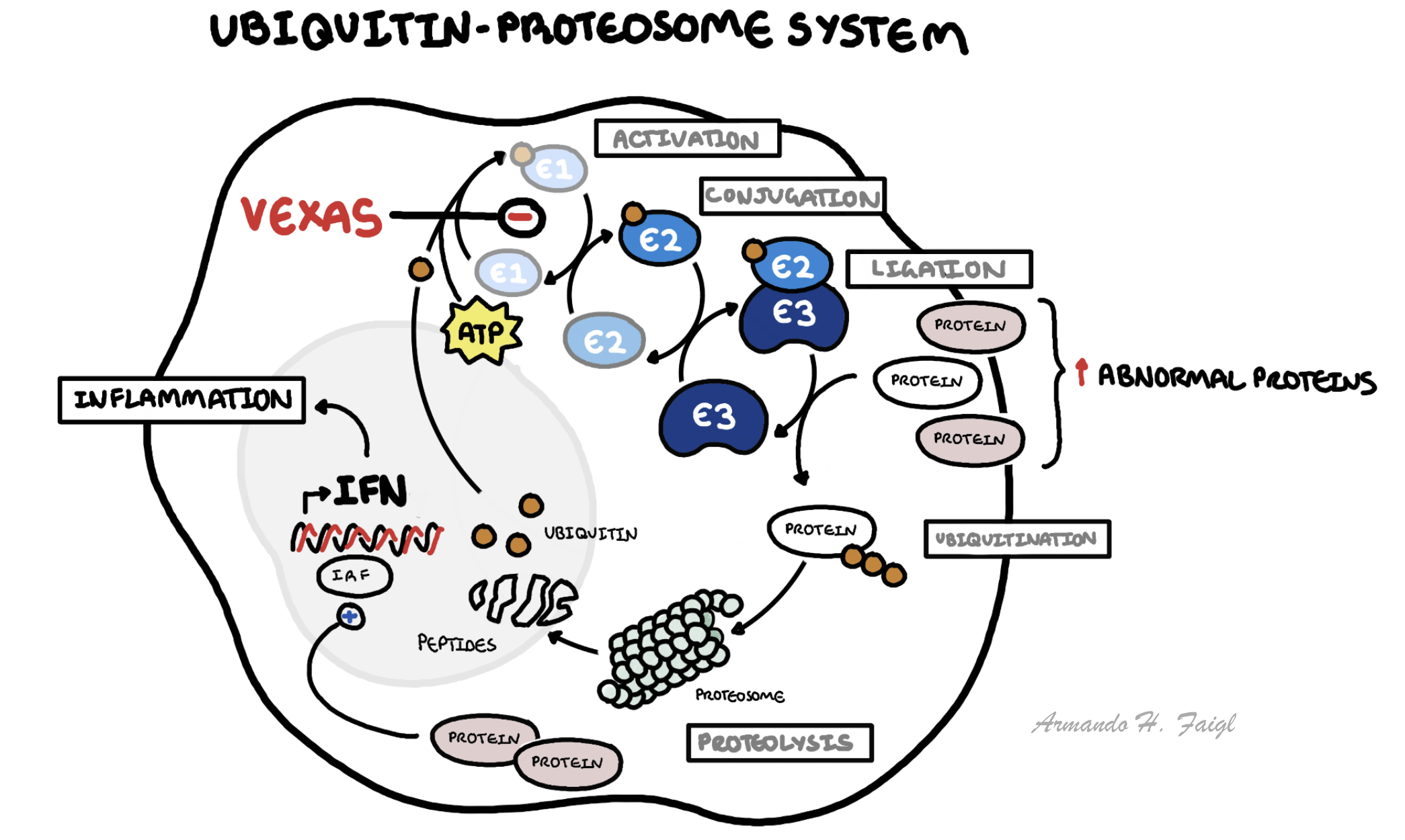
- Ubiquitin–proteasome system: Regulates protein degradation, cell signalling, and immune responses via tagging proteins for destruction.
- Bone marrow: Site of haematopoiesis; abnormalities can lead to cytopenias and dysplastic changes.
- Innate immune system: Includes macrophages, neutrophils, and cytokines (IL-1, IL-6, TNF-α), responsible for rapid inflammatory responses.
Aetiology & Risk Factors
Aetiology
- Somatic mutation in UBA1 gene in haematopoietic progenitor cells
- Leads to defective ubiquitination and dysregulated immune signalling
- Male sex (X-linked gene)
- Age >50 years
- Presence of clonal haematologic disorders (e.g., MDS, monoclonal gammopathy)
Always consider VEXAS in older men with refractory systemic inflammation + cytopenias.
- Somatic UBA1 mutation → defective ubiquitin activation
- Impaired protein degradation → accumulation of abnormal proteins
- Activation of innate immune pathways → cytokine overproduction (IL-6, TNF)
- Expansion of mutant myeloid clones → bone marrow dysfunction
- Systemic inflammation + haematologic abnormalities
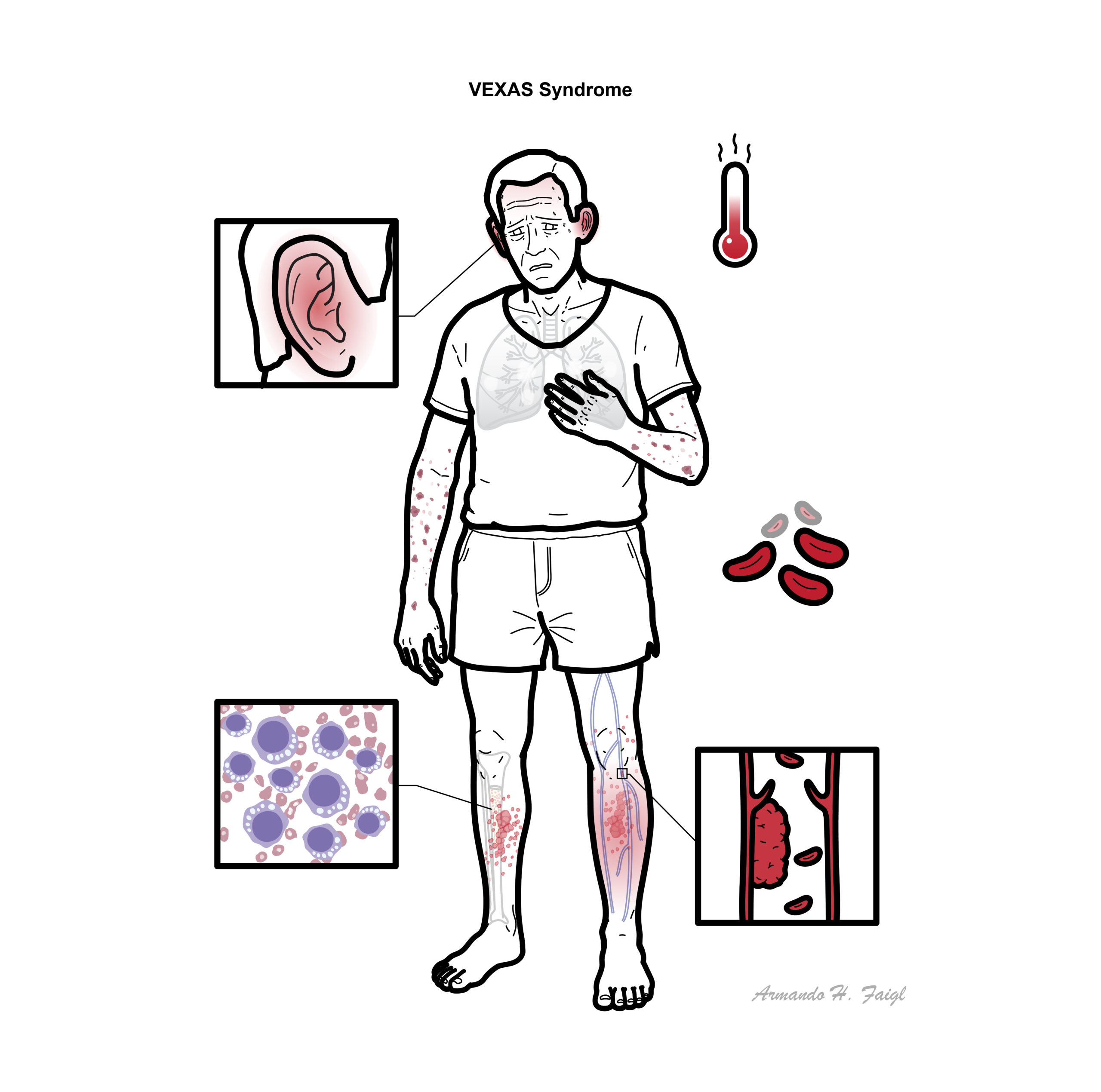
Clinical Manifestations
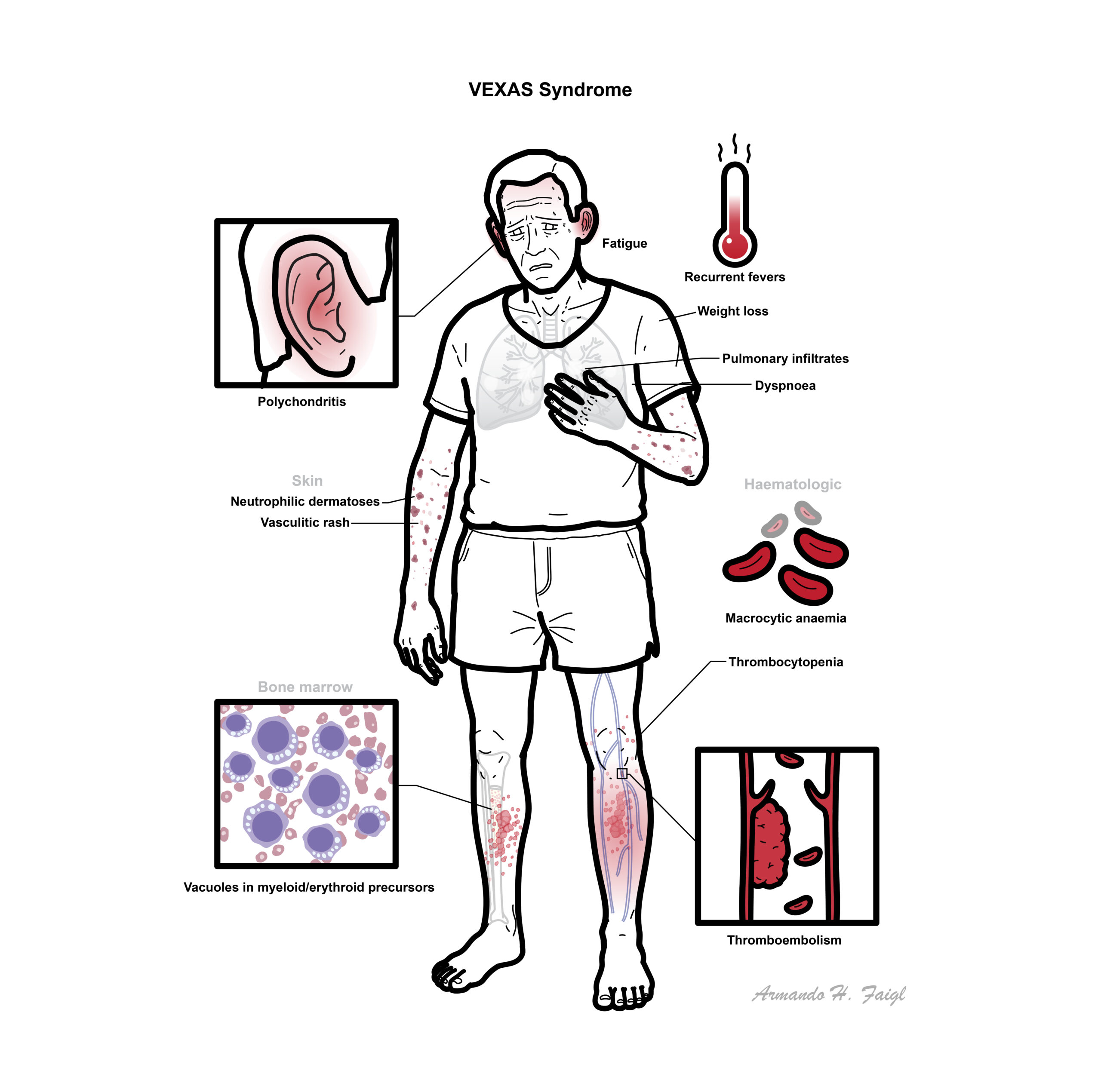
- Recurrent fevers, weight loss, fatigue
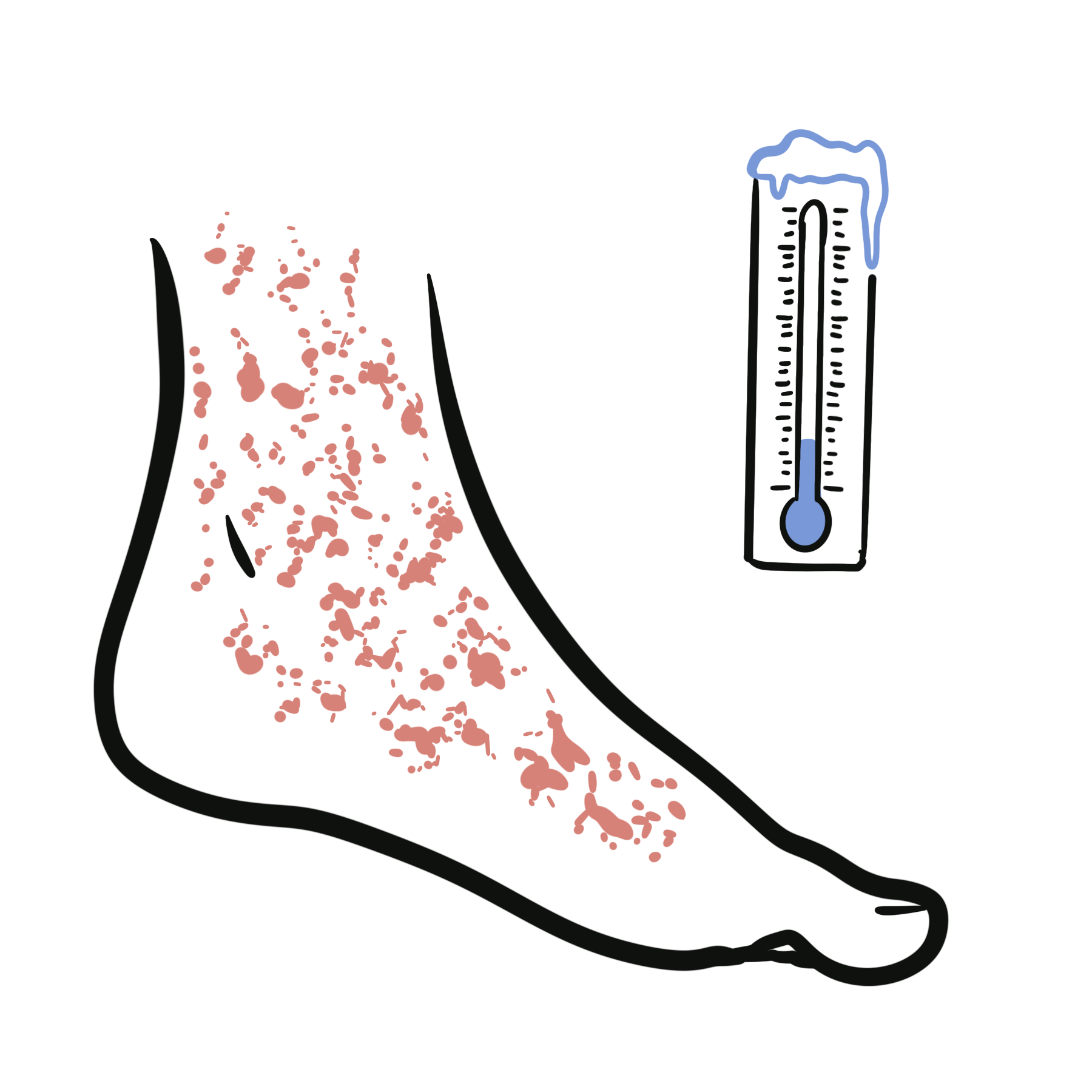
- Skin: neutrophilic dermatoses, vasculitic rash
- Pulmonary: infiltrates, dyspnoea
- Cartilage inflammation: relapsing polychondritis-like features
- Haematologic: macrocytic anaemia, thrombocytopenia
- Thromboembolism (high risk)
- Bone marrow: vacuoles in myeloid/erythroid precursors
Diagnosis
Diagnostic Criteria
- Clinical suspicion (older male + systemic inflammation + cytopenias)
- Bone marrow biopsy: vacuoles in myeloid/erythroid precursors
- Genetic testing: UBA1 mutation (confirmatory)
- FBC: macrocytic anaemia ± thrombocytopenia
- CRP/ESR: markedly elevated
- Bone marrow biopsy
- Genetic testing (UBA1 sequencing)
Differentials
- Myelodysplastic syndrome
- Relapsing polychondritis
- Adult-onset Still’s disease
- ANCA-associated vasculitis
Diagnosis requires genetic confirmation: UBA1 mutation.
Treatment
- Glucocorticoids: mainstay for symptom control (often steroid-dependent)
- Immunosuppressants: limited efficacy (methotrexate, azathioprine)
- Biologics: variable response (IL-6 inhibitors, JAK inhibitors promising)
- Haematologic therapies:
- Azacitidine for MDS-associated disease
- Allogeneic stem cell transplant (potentially curative in selected patients)
Complications & Prognosis
- Myelodysplastic syndrome progression
- Severe infections (due to immunosuppression)
- Thromboembolic events
- Organ damage (lung, skin, cartilage)
- Chronic, relapsing course
- High morbidity and mortality if untreated
- Prognosis worse with:
- Severe cytopenias
- Underlying haematologic malignancy
- Refractory inflammation
References
- Beck DB, Ferrada MA, Sikora KA, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. 2020;383(27):2628–2638.
- Obiorah IE, Patel BA, Groarke EM, et al. Benign and malignant hematologic manifestations in patients with VEXAS syndrome. Blood Adv. 2021;5(16):3206–3215.
- Georgin-Lavialle S, Terrier B, Guedon AF, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series. Lancet Rheumatol. 2022;4(3):e192–e202.
- Ferrada MA, Sikora KA, Luo Y, et al. Somatic mutations in UBA1 define a distinct subset of relapsing polychondritis patients with VEXAS. Arthritis Rheumatol. 2021;73(10):1886–1895.
- Poulter JA, Collins JC, Cargo C, et al. Novel somatic mutations in UBA1 as a cause of VEXAS syndrome. Blood. 2021;137(26):3676–3681.
- Bourbon E, Heiblig M, Gerfaud-Valentin M, et al. Therapeutic options in VEXAS syndrome: insights from a retrospective study. Blood. 2021;137(26):3682–3684.
- Comont T, Heiblig M, Rivière E, et al. Azacitidine for patients with VEXAS syndrome. Br J Haematol. 2022;196(4):969–974.
- Goyal G, Lacy MQ, Dispenzieri A, et al. Clinical characteristics and outcomes of patients with VEXAS syndrome. Blood Cancer J. 2022;12(3):54.
- van der Made CI, Potjewijd J, Hoogstins A, et al. Adult-onset autoinflammation caused by somatic mutations in UBA1: a review of VEXAS syndrome. J Clin Immunol. 2022;42(2):214–227.
- Patel BA, Groarke EM, Ferrada MA, et al. Thrombotic events in VEXAS syndrome: clinical features and outcomes. Blood. 2021;138(Supplement 1):3619.






Members only discussions coming soon…