“Each patient ought to feel somewhat the better after the physician’s visit irrespective of the nature of the illness.”
-Warfield Theobald Longcope
Overview
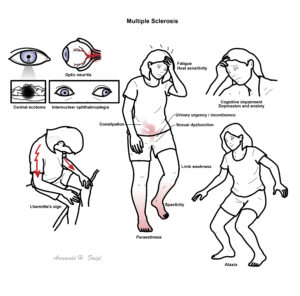
Multiple sclerosis (MS) is defined as an inflammatory demyelinating disease characterised by the presence of episodic neurological dysfunction in at least 2 areas of the CNS (brain, spinal cord, and optic nerves) separated in time and space.
MS is the most common cause of neurological disability among young adults. MS is most commonly diagnosed between 20 to 40 years old. Irreversible disability can occur, but life expectancy is generally not affected.
Definition
Multiple Sclerosis: Incurable disease of the central nervous system that can affect the brain, spinal cord and optic nerves. Guillain–Barré syndrome: Rapid-onset muscle weakness caused by the immune system damaging the peripheral nervous system. Motor Neuron Disease: Motor neurone disease is largely a sporadic disease of middle and elderly life presenting in the sixth and seventh decades. The classic form of the disease is also referred to as amyotrophic lateral sclerosis and presents with a mixture of upper and lower motor neurone features, such as wasted fasciculating biceps with a brisk or easily obtained biceps deep tendon reflex. The rarer variants of the disease can have a pure upper motor neurone presentation, primary lateral sclerosis, or a pure lower motor neurone presentation, progressive muscular atrophy. Myasthenia Graves:Autoimmune disease characterized by muscle weakness that fluctuates, worsening with exertion, and improving with rest. In about two-thirds of the patients, the involvement of extrinsic ocular muscle presents as the initial symptom, usually progressing to involve other bulbar muscles and limb musculature, resulting in generalized myasthenia gravis.
An immune mechanism is suggested by increased levels of activated T-lymphocytes in the CSF and increased Ig synthesis within the CNS
An attack CNS inflammation starts with the entry of activated T-lymphocytes across the BBB
T cells seek entry into the CNS via attachment to a receptor on endothelial cells
This interaction allows a breach in the BBB, leading to further upregulation of endothelial adhesion molecules and addition influx of inflammatory cells
These recognise myelin-derived antigens on the surface of nervous system’s APCs (the migroglia) and undergo clonal proliferation
The resulting inflammatory cascade releases cytokines and initiated destruction of oligodendrocyte-myelin unit by macrophages (oligodendrocytes try and repair the myelin but overtime as the myelin keeps getting destroyed, they become less effective at doing so)
The degernative component of MS is believed to reflect axonal degeneration and loss
Demyelination disrupts axonal support and leads to destabilisation of axonal membrane potentials which causes distal and retrograde degeneration over time (+ injury of the inflammatory cells to axons)
Course of disease
85% present with relapsing/remitting disease (RRMS)
Subacute evolution of symptoms over days; symptoms reach a plateau and resolve over days or weeks
After 10–15 years, 50% enter the secondary progressive phase some with relapses (SPMS).
10% have primary progressive disease (PPMS) with gradual accumulation of disability
Clinical Classification of MS
TYPES OF MULTIPLE SCLEROSIS BASED ON DISEASE ONSET
Relapsing remitting (85% of onset)
Discrete attacks that evolve over days to weeks, followed by some degree of recovery over weeks to months; the patient has no worsening of neurologic function between attacks
Primary progressive (10% of onset)
Gradual progression without acute attacks or improvements
Secondary progressive (50% of Relapsing remitting may evolve to secondary progressive)
Initial relapsing remitting disease, followed by gradual neurologic deterioration not associated with acute attacks
Relapsing progressive (5% of onset)
Like primary progressive, but with some superimposed relapses
TYPES OF MULTIPLE SCLEROSIS BASED ON DISEASE COURSE
Clinically isolated syndrome (commonest first presentation of multiple sclerosis)
Probable first episode of relapsing-remitting multiple sclerosis (eg optic neuritis, transverse myelitis or brainstem episode). If two or more white matter lesions on MRI then about 80% chance of eventual multiple sclerosis; if MRI is normal then only about 20% chance
Diagnosis Multiple sclerosis remains a clinical diagnosis supported by:
Magnetic resonance imaging (MRI)
Evoked potential studies (delayed evoked response with preserved waveform)
Cerebrospinal fluid examination (oligoclonal bands and raised IgG index)
Diagnosis
Clinical: two neurologic deficits (e.g., focal weakness, sensory disturbances) separated in time and space, in the absence of fever, infection, or competing etiologies, are considered diagnostic
Attacks may be patient-reported or objectively observed, and must last for a minimum of 24 hours.
MRI – Diagnosing MS in cases not meeting the threshold for clinical diagnosis
Evoked potentials (visual, auditory, and somatosensory) may provide objective evidence of deficits consistent with MS
Cerebrospinal fluid – Oligoclonal bands +ve in CSF but NOT in serum, Increased CSF IgG synthesis.
Typical sites of lesions on MRI
Periventricular white matter (if at right angles to the corpus callosum, these are referred to as ‘Dawson fingers’)
Juxtacortical white matter
Corpus callosum
Optic nerve
Infratentorial structures (pons, cerebellar peduncles and cerebellum)
Spinal cord
Treatment
With symptoms of relapse, it is vital to establish whether the symptoms are definitely neurological. Intercurrent infection, fever, increased environmental temperature, heightened body awareness and depression can all produce ‘pseudorelapses’. Many ‘attacks’ simply reflect heightened body awareness and anxiety, particularly in the first year after diagnosis.
Immunosuppressants and newer therapies (second line)
Life style modifications (for urinary frequency and motor problems)
Avoid caffeine and vitamin waters
Yoga and relaxation
Physiotherapy excercise
Pharmacology
Alemtuzumab is a humanised monoclonal Ab binds CD52 (on B cells). Live vaccines are contraindicated. Side effects: Good pastures, 2-3% ITP, 20-30 % autoimmune thyroid disease at 5 years.
Pharmacology
Natalizumab Monoclonal antibody that works by selective inhibition of adhesion molecules, slowing entry of T cells through cerebral capillaries. Used in patients intolerant of/not responding to other immunomodulators. Side effects: fatigue, hypersensitivity hepatotoxicity and has been associated with progressivemultifocal leukoencephalopathy.
~25% of patients with multiple sclerosis have a benign course throughout, with minimal or no disability after many years.
The majority of patients have a relapsing-remitting course of disease at the time of initial diagnosis
After 10 years, about half of the patients with relapsing-remitting disease will have changed to a progressive form (secondary progressive); this transition worsens the prognosis.
Progressive multifocal leucoencephalopathy (PML)
Rapidly progressive demyelination in brain
Characterised by dementia, motor dysfunction and visual loss
JC = John Cunningham virus (polyoma virus) infection of oligodendrocytes causes PML
Diagnosis: JC virus in CSF via PCR
PML occurs in immunocompromised (AIDS, transplant Pt, immune system altering drugs e.g., MS Patient on disease modifying therapy)
Most effective Rx is immune reconstitution
IRIS = immune reconstitution syndrome
Neuromyelitis optica (Devic disease)
Antibody-mediated inflammation directed at aquaporin-4 channels in the CNS that results in inflammatory demyelination in the optic nerves and spinal cord
Can be differentiated from MS by
NMO IgG antibody testing
Lack of significant brain involvement, large and longitudinally extensive spinal cord lesions
Doesn’t respond to interferon or other MS treatments
Treatment is immunosuppression – steroids, rituximab.
Worse prognosis than MS. – 50% walking problems at 5 years
Acute Disseminated Encephalomyelitis (ADEM)
Occurs in children
ADEM is an acute demyelinating disorder. It is thought to be an autoimmune response to myelin basic protein as a result of a viral illness or vaccination.
ADEM presents with a short history of flu-like illness, with subsequent development of focal neurology, fever and encephalopathy
Treatment is usually with intravenous steroids
It is a differential for multiple sclerosis
all the lesions are gadolinium enhancing whereas in multiple sclerosis some lesions might not enhance
References
UpToDate Best Practice BMJ AAFP – Multiple Sclerosis: A Primary Care Perspective RACGP – Multiple Sclerosis: Diagnosis, management and prognosis
















Discussion