

Chronic Spontaneous Urticaria +/- angioedema

Overview
Chronic spontaneous urticaria (CSU) is a mast-cell–driven skin disorder defined by recurrent wheals (hives), angioedema, or both for >6 weeks without a specific external trigger, with a point prevalence around 0.5–1% and a female predominance; median disease duration is ~2–5 years, but a subset persists longer and markedly impairs quality of life and work productivity. Autoimmune mechanisms (type I “autoallergic” IgE-mediated and type IIb IgG-autoantibody–mediated) account for a large fraction of cases, and comorbidities include atopy, autoimmune thyroid disease, and anxiety/depression.
Definition
CSU: spontaneous wheals, angioedema, or both, on most days for >6 weeks, without a consistent external trigger.
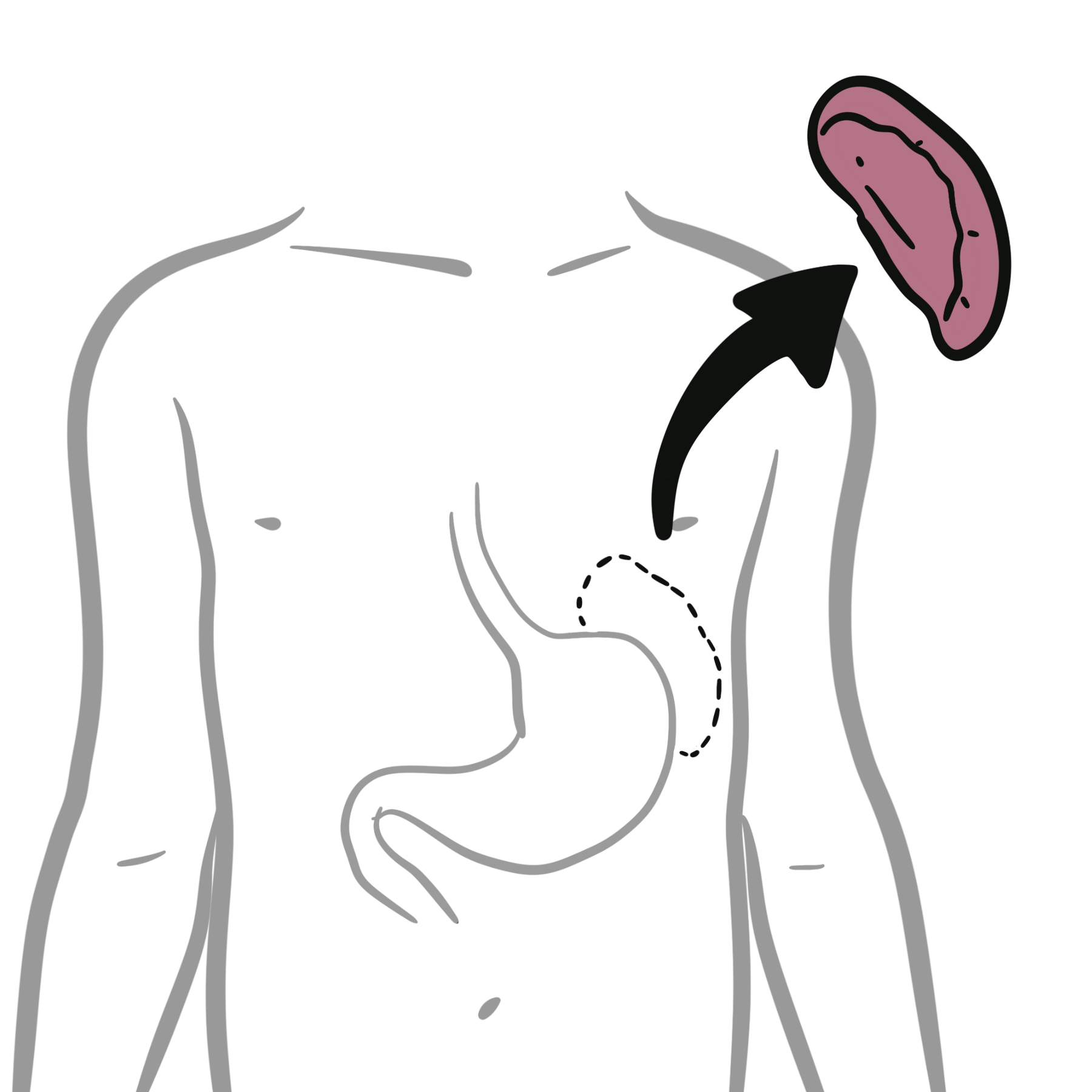
Angioedema (histaminergic): transient, deeper swelling (face, lips, tongue, genitalia), often painful/tingly, usually with or without wheals in CSU.
Triad Daily itchy fleeting wheals, ± angioedema, >6 weeks with no consistent trigger.
Aetiology & Risk factors
Aetiology
- Autoimmune mechanisms: type I autoallergic (IgE to self-antigens) and type IIb autoimmunity (anti-FcεRIα/anti-IgE) with basophil activation and low total IgE
- Non-specific mast-cell hyper-reactivity: neuropeptides (substance P), MRGPRX2 agonists, innate signals.
- Female sex
- Middle age onset
- Personal/family atopy
- Autoimmune thyroid disease and other autoimmunity
- NSAID-exacerbated cutaneous disease; stress, infections; obesity and metabolic syndrome reported in some cohorts
Low total IgE, high CRP/ESR, positive BAT/ASST, thyroid autoimmunity → suspect type IIb phenotype with higher refractoriness to antihistamines and better response to cyclosporine than omalizumab.
Pathophysiology
- In chronic urticaria, mast cells and basophils become abnormally sensitive and overactive, often due to autoimmune mechanisms.
- In some patients, IgE autoantibodies bind self-antigens, while in others IgG autoantibodies target FcεRI (the high-affinity IgE receptor) or IgE itself on mast cells and basophils.
- This leads to activation of these cells through FcεRI cross-linking or related immune pathways, sometimes with contribution from complement.
- Activated mast cells and basophils then release histamine, tryptase, leukotrienes, prostaglandins, and other inflammatory mediators.
- Histamine acts on H1 and H2 receptors in the skin vasculature, causing vasodilation, increased vascular permeability, and sensory nerve stimulation, which produce erythema, edema, and itch.
- When this vascular leakage occurs in the superficial dermis, it causes urticarial wheals; when it occurs in the deeper dermis and subcutaneous tissue, it causes angioedema.
- The inflammatory response is further amplified by cytokines such as IL-4, IL-5, IL-13, IL-31, and TNF, which promote ongoing immune activation and increase mast-cell responsiveness.
- Neuropeptides and signals from the coagulation/fibrinolytic system may also contribute, helping explain why some patients have more severe or persistent disease, sometimes with an elevated D-dimer.
Histamine drives itch and edema → explains the central role of H1-antihistamines; persistent activity implicates autoimmunity in many patients.
Clinical Manifestations
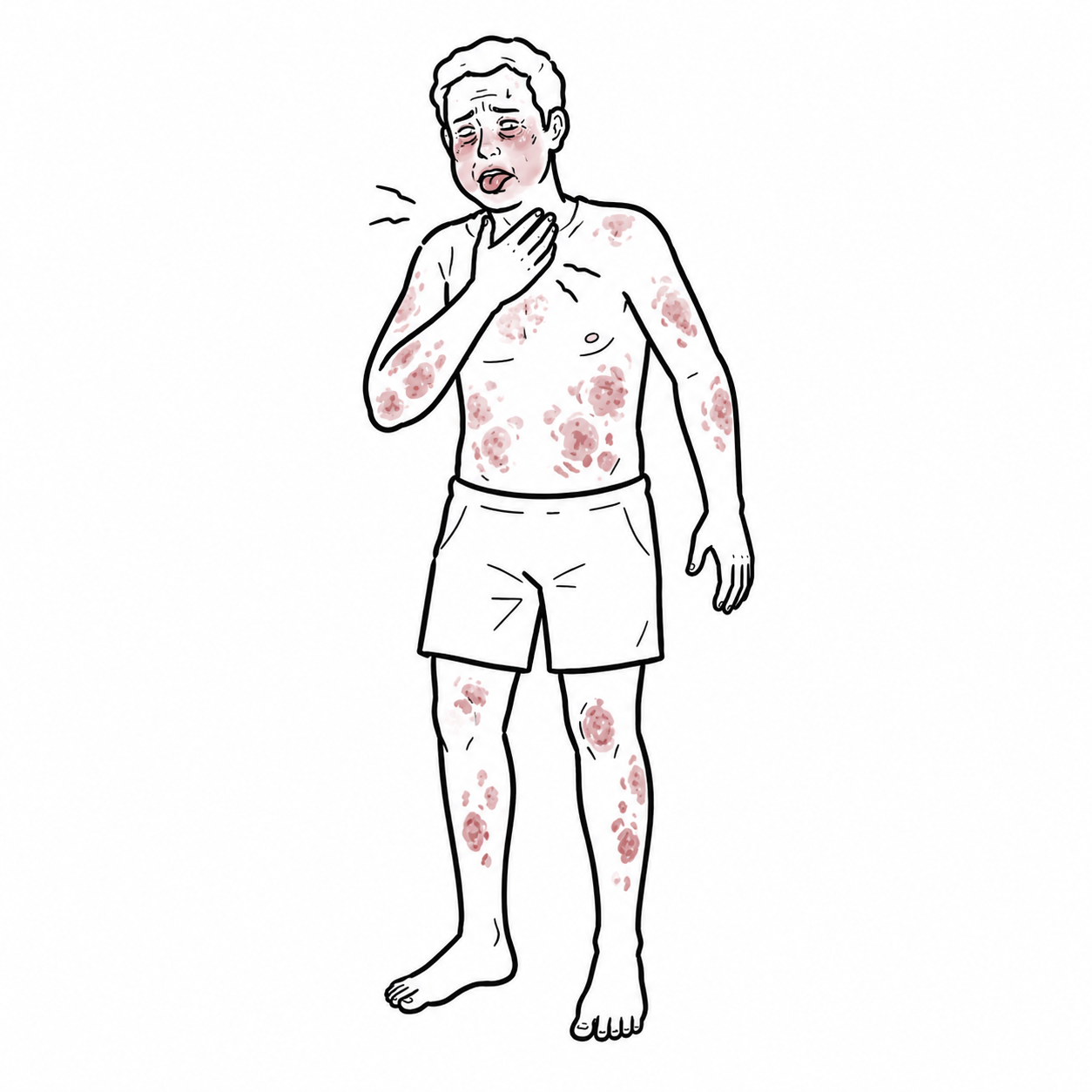
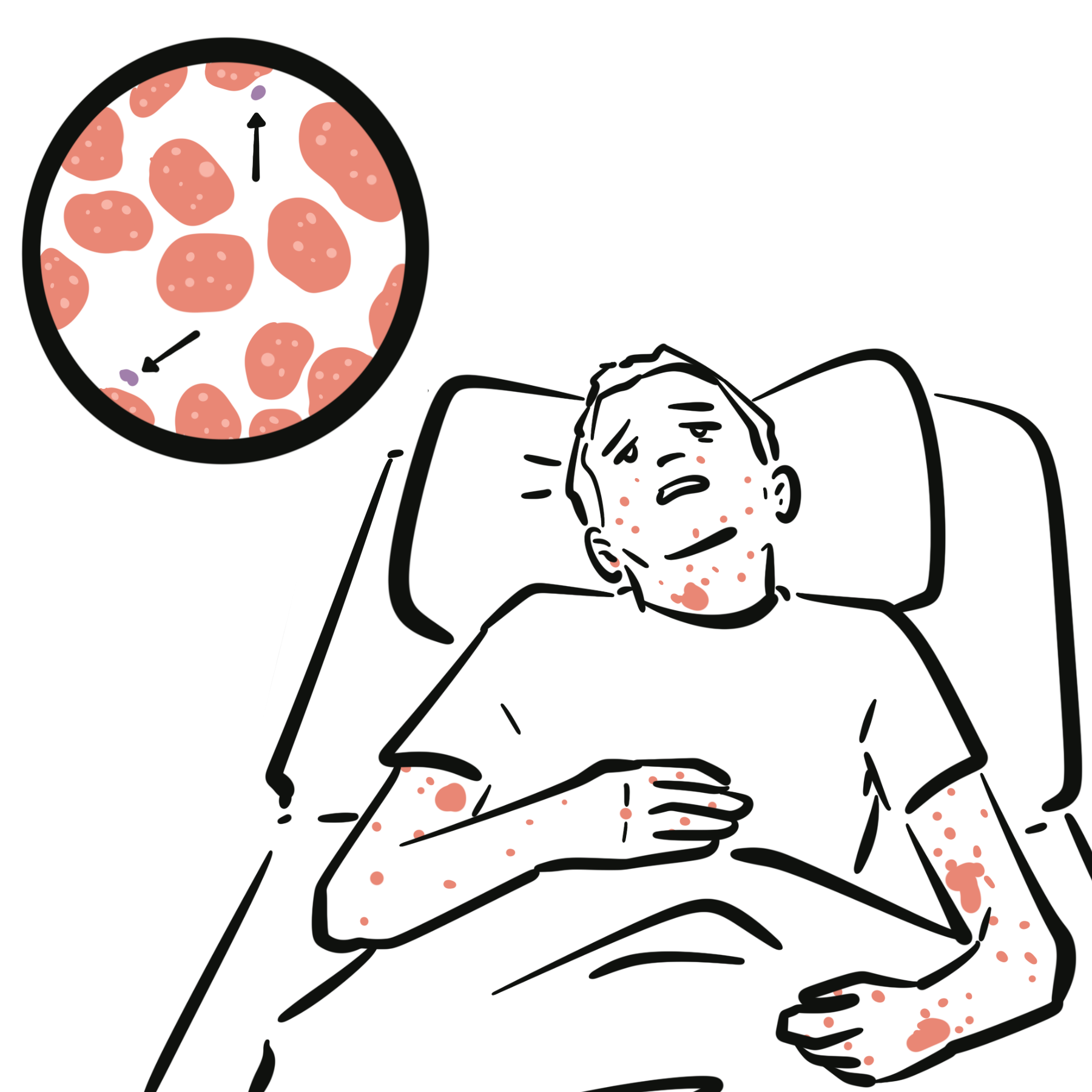
- Intensely pruritic wheals of variable size/shape, migrating, each lesion <24 h
- Angioedema in ~40–50%.
- Course: daily or near-daily flares >6 weeks
- Symptoms are worse at night causing sleep disturbance, anxiety/depression, work/school impairment.
- Triggers/exacerbators (not causal): NSAIDs, stress, heat/pressure/tight clothing, infections; some foods may exacerbate but no consistent allergen like in acute urticaria
Diagnosis
Clinical diagnosis: recurrent wheals/angioedema on most days for >6 weeks without consistent external trigger; exclude differentials
- Chronic inducible urticarias: reproducible by specific stimuli (cold, heat, pressure, cholinergic, solar) → confirm with provocation tests
- Urticarial vasculitis: lesions >24–48 h, painful/burning, bruising/hyperpigmentation, raised CRP/ESR; biopsy with leukocytoclastic vasculitis
- Bradykinin-mediated angioedema (ACE-i/HAE): angioedema without itch/wheals, prolonged, antihistamine/steroid-resistant
- Mastocytosis/MCAS: recurrent flushing, systemic mediator symptoms, elevated baseline tryptase
Lesions persisting >24–48 h, painful with bruising or residual hyperpigmentation → consider urticarial vasculitis and biopsy..
Do not over-investigate. Most CSU needs minimal labs.
Treatment
- Education & general measures
- reassure chronic but benign course
- avoid NSAIDs (if exacerbating)
- heat/tight clothing
- optimise sleep/stress
- trigger diaries
- Step 1 — First-line: Second-generation, non-sedating H1-antihistamine daily (cetirizine, levocetirizine, fexofenadine, bilastine, loratadine, desloratadine) at standard dose
- Step 2 — Up-dosing: if uncontrolled after 2–4 weeks, increase to as much as 4-fold the standard dose (e.g., fexofenadine up to 720–960 mg/day in divided doses) within label/regional guidance; avoid chronic first-generation agents due to anticholinergic/CNS effects
- Step 3 — Add biologic: Omalizumab 300 mg SC q4w (some patients respond to 150 mg); up-dosing or interval shortening (e.g., 300–600 mg q2–4w) for partial responders under specialist care
- Step 4 — Add immunosuppressant: Cyclosporine (≈3–5 mg/kg/day) when omalizumab fails/contraindicated, with BP/renal monitoring; often effective in type IIb endotype
- Rescue for severe flares: short oral corticosteroid burst (e.g., ≤10 days) may be used sparingly; avoid repeated/long courses
- Emerging/near-future: BTK inhibitors (e.g., remibrutinib) show promising phase-3 efficacy; ligelizumab has mixed phase-3 results; dupilumab shows benefit in trials/real-world cohorts for antihistamine-refractory CSU but guideline positioning varies by jurisdiction.
Complications & Prognosis
- Sleep disturbance
- Anxiety/depression
- Presenteeism/absenteeism; steroid toxicity if mismanaged; ED visits for angioedema flares.
Prognosis
~50% remit within 1–3 years; many within 5 years, but a subset persists longer; type IIb, angioedema-predominant, high baseline activity (UAS7), elevated CRP/ESR, and low total IgE predict more severe/refractory disease.
References
- Zuberbier T, Abdul Latiff AH, Abuzakouk M, et al. The international EAACI/GA²LEN/EuroGuiDerm/APAAACI guideline for urticaria—2021 update and revision. Allergy. 2022;77(3):734–766.
- Zuberbier T, Maurer M, Weller K, et al. Definition, classification, diagnosis, and management of chronic urticaria: A GA²LEN position paper. Allergy. 2018;73(7):1393–1414.
- Sánchez-Borges M, Ansotegui IJ, Baiardini I, et al. Diagnosis and treatment of urticaria and angioedema: 2022 WAO/EAACI consensus. World Allergy Organ J. 2022;15(12):100733.
- Sussman G, Hébert J, Gulliver W, et al. Real-world burden and epidemiology of chronic spontaneous urticaria. J Cutan Med Surg. 2018;22(5):427–436.
- Kolkhir P, Altrichter S, Hawro T, Maurer M. Autoimmune chronic spontaneous urticaria. J Allergy Clin Immunol. 2021;147(6):1865–1878.
- Schoepke N, Asero R, Ellrich A, et al. Biomarkers and clinical characteristics of type IIb autoimmune CSU. Allergy. 2019;74(12):2427–2436.
- Bae YJ, Shin YS, Park HS. Clinical implications of basophil tests in chronic urticaria. Curr Opin Allergy Clin Immunol. 2020;20(5):417–424.
- Sabroe RA, Greaves MW. Histamine and urticaria: an appraisal. Clin Exp Dermatol. 2019;44(8):875–882.
- Sánchez Díaz M, Curto Barredo L, Ferrer M. Comorbidities and triggers in CSU. Immunol Allergy Clin North Am. 2022;42(1):101–117.
- Busse PJ, Christiansen SC. Hereditary and acquired angioedema. N Engl J Med. 2020;382(12):1136–1148.
- Bernstein JA, Lang DM, Khan DA, Craig T. Updates in CSU management: antihistamines, omalizumab, cyclosporine, and beyond. J Allergy Clin Immunol Pract. 2023;11(1):22–34.
- Saini SS, Kaplan A, Maurer M. Current and emerging biologics in CSU. J Allergy Clin Immunol. 2021;148(5):1173–1181.
- Maurer M, Giménez-Arnau A, Godek D, et al. BTK inhibitors and novel biologics for CSU—evidence and positioning. Allergy. 2024;79(5):1099–1112.













Members only discussions coming soon…