Overview
Immunoglobulin A vasculitis (IgAV), also known as Henoch–Schönlein purpura, is the most common primary vasculitis in children (peak incidence 4–7 years), with seasonal peaks after upper respiratory tract infections. It is defined as a small-vessel vasculitis with IgA1-dominant immune deposits affecting capillaries, venules, and arterioles, underscoring the central role of IgA in its pathogenesis. Clinically, IgAV can be divided into two forms: a skin-limited form, characterised by cutaneous IgA-dominant vasculitis without systemic organ involvement and manifesting solely as non-thrombocytopenic purpura, and a systemic form, where skin involvement is accompanied by at least one other organ, most commonly the joints, gastrointestinal tract, and kidneys, and rarely the lungs or central nervous system.
The disease is often self-limiting, but recurrence occurs in around one-third of paediatric cases. Renal involvement is the most important determinant of prognosis. Between 20% and 80% of children develop nephritis within four to six weeks of presentation, and 1–7% progress to renal failure or end-stage renal disease (ESRD). IgA vasculitis is milder in children younger than two years, but more severe in adults, with worse outcomes.
Definition
Galactose-deficient IgA1 (Gd-IgA1): Aberrantly O-glycosylated IgA1 subclass prone to forming nephritogenic immune complexes.
IgAV nephritis (IgAVN): Kidney involvement in IgA vasculitis, marked by IgA1-dominant immune complex deposition in the glomerular mesangium, leading to haematuria, proteinuria, and variable renal impairment within a systemic small-vessel vasculitis.
IgA nephropathy (IgAN): chronic glomerular disease characterised by IgA1-dominant immune complex deposition in the mesangium, causing haematuria, proteinuria, and potential progression to kidney failure. No vasculitis present.
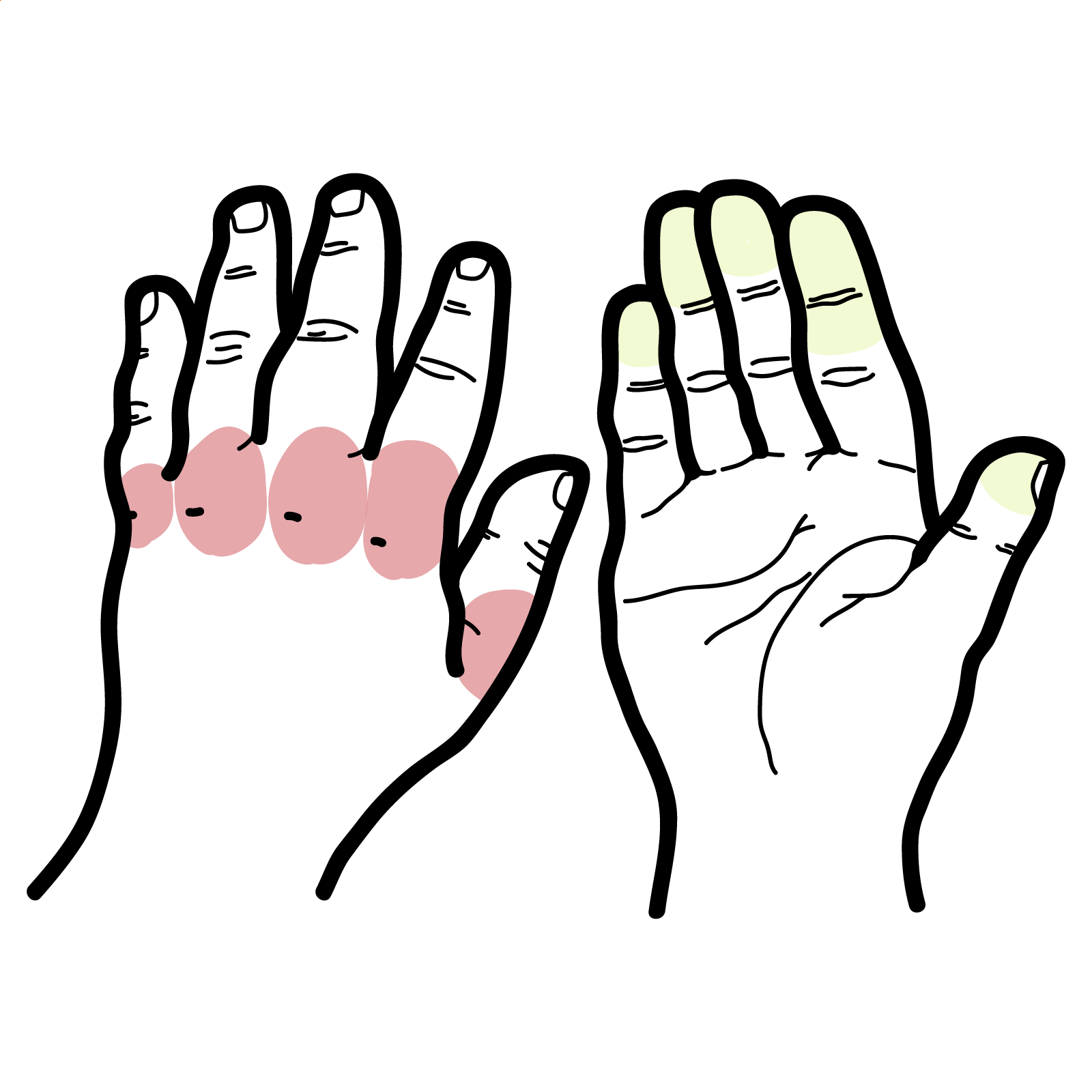
Palpable purpura: Non-blanching, raised purpuric lesions from leukocytoclastic vasculitis of dermal venules.
Remember
Adults = lower incidence, higher renal risk; always monitor BP, urinalysis, and proteinuria for ≥6–12 months [4,5].
Aetiology & Risk Factors
Aetiology
- Post-infectious mucosal immune dysregulation (commonly URTI) → increased Gd-IgA1 ± anti-Gd-IgA1 autoantibodies → immune complex deposition
- Genetic predisposition
Risk Factors for Severe/Renal Disease
- Older age (especially adults)
- Male sex
- Persistent proteinuria
- Hypertension
- Reduced GFR at onset
Think
Early proteinuria (>0.5–1 g/day) and hypertension are actionable — start RAAS blockade promptly.
Pathophysiology
IgA vasculitis (IgAV) is a leukocytoclastic vasculitis characterised by IgA-dominant immune complex deposition in small vessels. Two main pathogenic models explain its phenotypes.
Anti-Gd-IgA1 autoantibodies: IgA vasculitis nephritis (IgAVN) mechanism parallels the “four-hit” theory of IgA nephropathy:
- Trigger (often mucosal infection) → overproduction of Gd-IgA1
- Formation of anti-Gd-IgA1 autoantibodies → circulating immune complexes
- Deposition in small vessels/mesangium → activation of alternative/lectin complement pathways
- FcαRI-mediated neutrophil recruitment → leukocytoclastic vasculitis in skin, bowel wall edema/bleeding, mesangial proliferation with proteinuria/hematuria [1,3,11]
Anti-endothelial cell antibodies (AECA)
- Neutrophil-driven small-vessel injury initiated by IgA1-type anti-endothelial cell antibodies (AECA) binding to β2 glycoprotein I receptors on endothelial cells
- Induces interleukin-8 production, neutrophil recruitment, and endothelial damage via antibody-dependent cellular cytotoxicity, complement activation, and reactive oxygen species.
Remember
Higher serum Gd-IgA1 correlates with extracutaneous/renal involvement in some patients [8,9].
Remember
Complement deposition pattern (IgA + C3, absent C1q) supports non-classical pathway activation.
Clinical Manifestations
Signs and symptoms may develop over days to weeks in any sequence. The onset of IgA vasculitis typically follows an upper respiratory infection. Joint involvement and abdominal pain are more common in children, whereas adults are more likely to have lower-extremity edema and hypertension
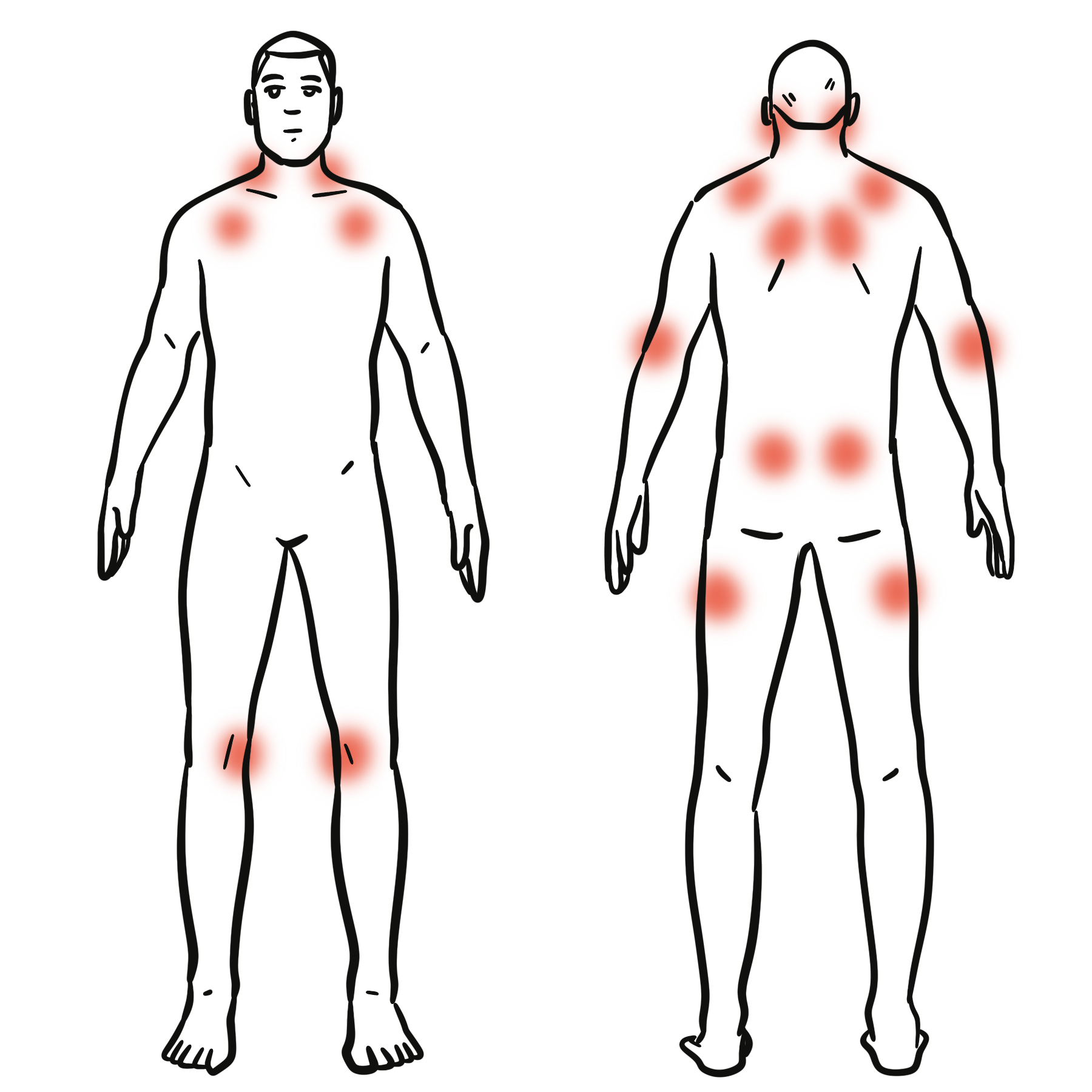
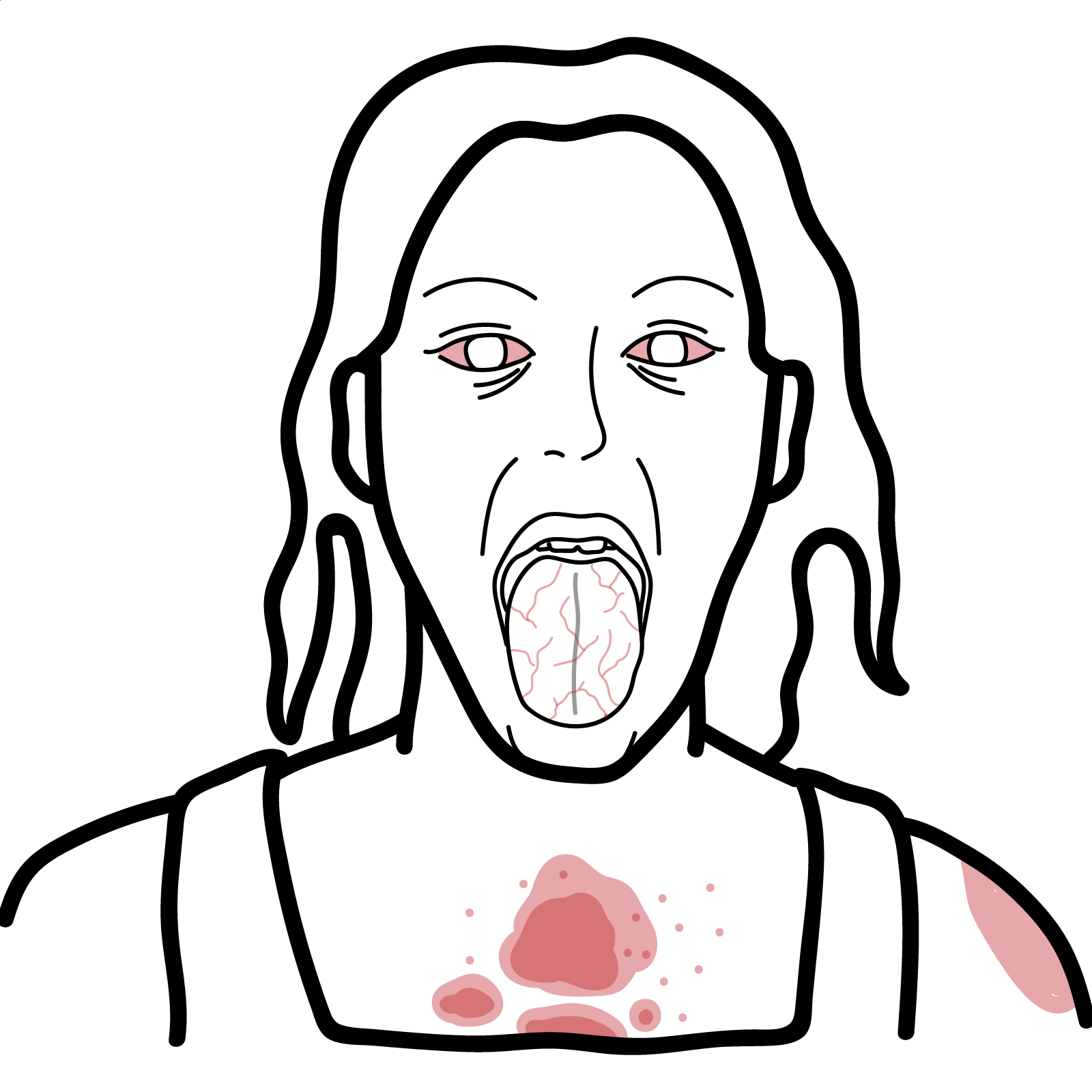
- Cutaneous: Palpable purpura (gravity-dependent; legs/buttocks ± arms/trunk), may form bullae or ulcerate in adults
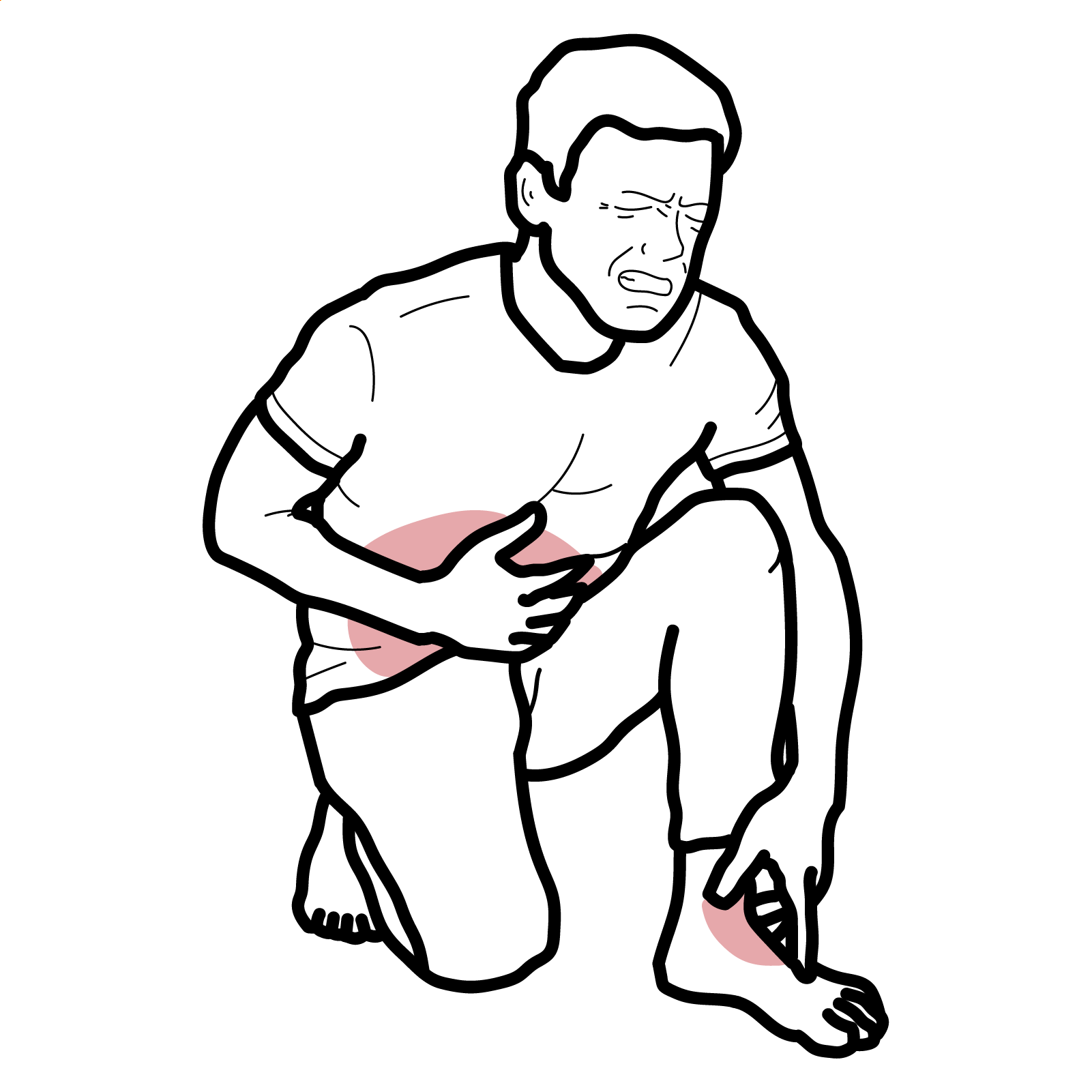
- Musculoskeletal: Arthralgia/arthritis (knees/ankles common), transient, non-erosive
- GI: Colicky abdominal pain, GI bleeding, intussusception (children), bowel ischemia/edema
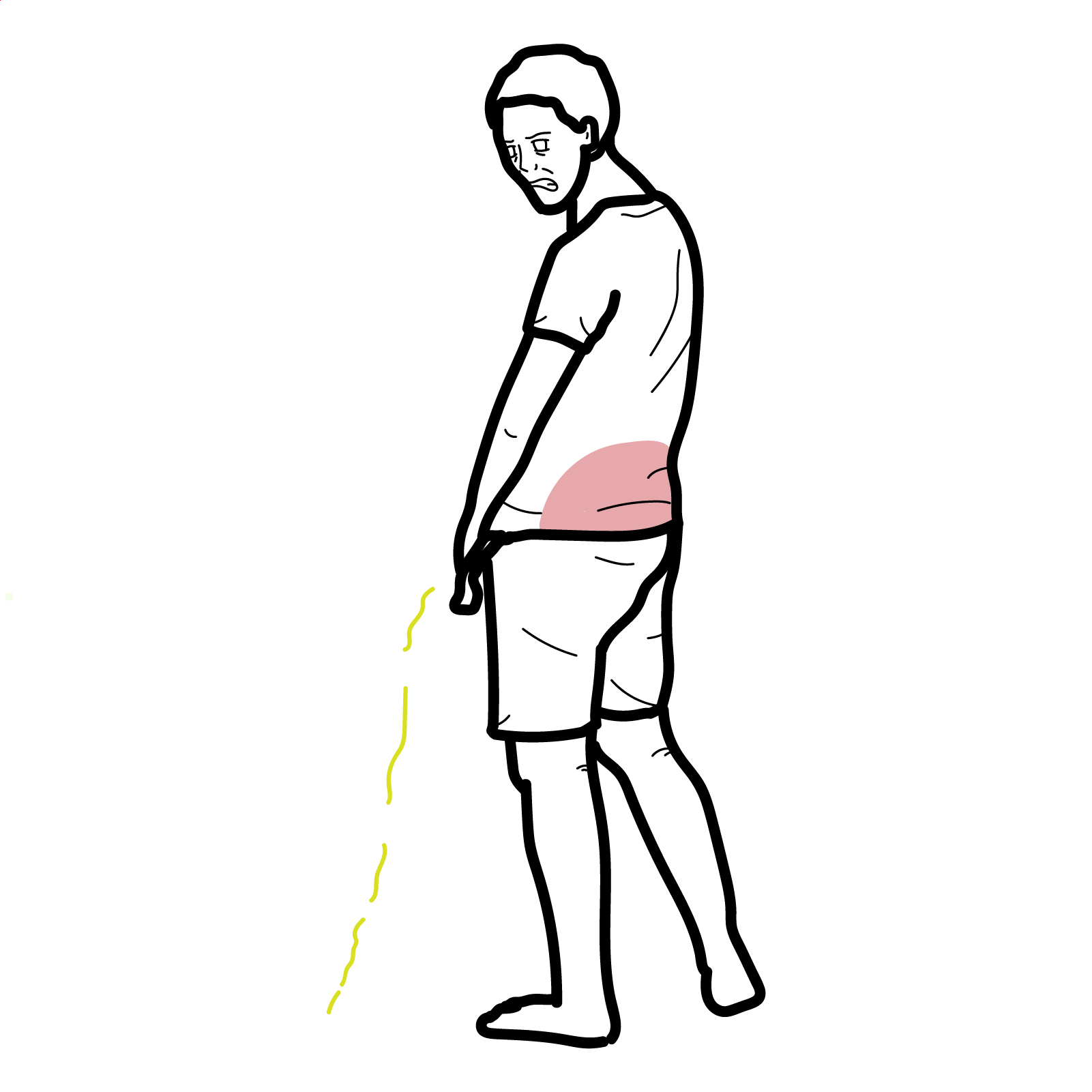
- Renal: Microscopic hematuria, proteinuria (range to nephrotic), hypertension, AKI
- Other: Scrotal edema, pulmonary hemorrhage
Remember
Always document BP and urinalysis — renal involvement may be silent but prognostically decisive.
Think
In adults with purpura + abdominal pain, check stool/occult blood and consider abdominal imaging before skin rash appear.
Tetrad: Palpable purpura + Arthralgia/arthritis + Abdominal pain/GI bleeding + Renal involvement [1].
Side note
Gastrointestinal symptoms at diagnosis are the best predictor of relapse in adults.
Diagnosis
EULAR/PRINTO/PRES classification criteria (2010)
- Mandatory: Purpura or petechiae
- Plus at least one of the following:
- Abdominal pain
- Arthritis or arthralgia
- Renal involvement
- Leukocytoclastic vasculitis with predominant IgA deposits or proliferative glomerulonephritis with predominant IgA deposits
Investigations
- Urinalysis (RBCs, protein), urine ACR/PCR
- Serum creatinine/eGFR
- Stool/occult blood if GI symptoms
- Abdominal US/CT for severe GI symptoms or suspected intussusception
- Renal biopsy if significant proteinuria, hematuria with casts, impaired GFR, or atypical course
- Skin biopsy for IgA-predominant leukocytoclastic vasculitis if uncertain
Differential Diagnosis
- ANCA-associated vasculitis (MPO/PR3+, pauci-immune GN)
- Cryoglobulinemic vasculitis (low C4, HCV+)
- SLE vasculitis (biopsy: “full house” IF, ANA+)
- Infection-related GN (low complement, subepithelial humps)
- Drug-induced leukocytoclastic vasculitis (no IgA dominance)
- IgA Nephropathy (IgAN)
| Feature | IgA Vasculitis Nephritis (IgAVN) | IgA Nephropathy (IgAN) |
| Primary disease | Systemic small-vessel vasculitis with renal involvement | Kidney-limited glomerular disease |
| Extrarenal features | Common – purpura, arthritis/arthralgia, abdominal pain, GI bleeding | Absent |
| Onset | Often acute, post-infectious, with rash ± systemic symptoms | Often insidious, detected via abnormal urinalysis |
| Age | Mostly children, can occur in adults (more severe course) | All ages; peak in teens–30s |
| Seasonality | Often seasonal (autumn/winter) | No seasonal pattern |
| Prognosis | Generally better; proteinuria may be transient | Worse long-term; high risk of CKD/ESKD over decades |
Classification
- Skin-limited IgAV
- Systemic IgAV without nephritis
- IgAV with nephritis (IgAVN) — most common in adults
Treatment
Supportive (all patients)
- Education, rest, analgesia, skin care
- Strict BP control
- RAAS blockade for proteinuria ≥0.5–1 g/day
- Avoid nephrotoxins
Glucocorticoids
- Not for prophylaxis of renal disease
- Indicated for severe GI involvement, scrotal edema, or established IgAVN [4,5]
IgAV Nephritis
- RAAS blockade ± glucocorticoids for moderate–severe proteinuria
- Cyclophosphamide for severe crescentic GN
- MMF, calcineurin inhibitors, rituximab in selected/refractory cases
Refractory Cutaneous Disease
- Dapsone as steroid-sparing; monitor Hb and methemoglobin
Complications & Prognosis
- Persistent proteinuria
- Hypertension
- CKD/ESKD (adults at higher risk)
- Gastrointestinal Haemorrhage, ischemia/perforation
- Intussusception (children),
- Relapses: Usually cutaneous-predominant
Prognosis
- IgAV spontaneously resolves in >90% of children and 90% of adults, making supportive treatment the primary intervention
- IgAV without nephritis is usually self-limiting, especially in children
- Higher IgM deposition in skin may indicate potential renal involvement
- IgAV with nephritis is associated with worse long-term prognosis. Not self-limiting — chronic inflammation can lead to permanent kidney damage
- Prognosis in children generally better than in adults. Adults have more severe and lasting kidney injury.
Remember
Children usually recover; adults need long-term renal surveillance.
References
- Yang Y, Sun L, Li G, et al. IgA vasculitis update: Epidemiology, pathogenesis, and biomarkers. Front Immunol. 2022;13:921864.
- Heineke MH, Ballering AV, Jamin A, Ben Mkaddem S, Monteiro RC, Van Egmond M. New insights in the pathogenesis of immunoglobulin A vasculitis (Henoch–Schönlein purpura). Semin Immunopathol. 2017;39(5):593-607.
- Liu LJ, Chen HP, Yu F, Zhao MH. Pathogenesis of IgA Vasculitis: An up-to-date review. Front Immunol. 2021;12:771619.
- The Royal Children’s Hospital Melbourne. Clinical Practice Guidelines: Henoch–Schönlein Purpura. 2021 (updated).
- KDIGO Glomerular Diseases Work Group. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int. 2021;100(4S):S1-S276.
- Hočevar A, Rotar Ž, Jurčić V, et al. IgA vasculitis in adults: performance of the EULAR/PRINTO/PRES classification criteria. Arthritis Res Ther. 2016;18:58.
- Bayindir Y, et al. Performance in adults of the EULAR/PReS/Ankara 2008 classification criteria for IgA vasculitis. RMD Open. 2025;11(3):e005728.
- Davin JC, Ten Berge IJ, Weening JJ. IgA nephropathy and IgA vasculitis with nephritis share Gd-IgA1-oriented pathogenesis. Kidney Int. 2017;91(3):534-536.
- Davin JC, Coppo R. The cause and pathogenesis of IgA nephropathy. Nat Rev Nephrol. 2014;10(6):339-352.
- Leone A, et al. Navigating adult-onset IgA vasculitis-associated nephritis. Life (Basel). 2024;14(8):930.
- Liu X, et al. Autoantibodies specific for Gd-IgA1 in IgA vasculitis nephritis. Kidney Int Rep. 2019;4(10):1553-1561.
- Song Y, et al. Dapsone in refractory IgA vasculitis: systematic review. BMC Nephrol. 2020;21:241.
- Kraitsidou E, et al. Adult-onset IgA vasculitis: update on therapy. Rev Med Interne. 2020;41(9):623-630.
- Thiel J. What’s new in treatment: IgA vasculitis & IgA nephropathy. The Rheumatologist. 2024.
- Glucocorticoids in IgA vasculitis. Acta Medica. 2024;65(4):e1100.



Discussion