
Overview
Systemic Lupus Erythematosus (SLE) is a chronic, multisystem autoimmune disease characterized by the production of autoantibodies and the deposition of immune complexes in various tissues. It predominantly affects women of childbearing age, with a female-to-male ratio of approximately 9:1. The disease is more prevalent among individuals of African, Asian, and Hispanic descent, and its overall prevalence ranges from 20 to 150 cases per 100,000 people, varying by geographic region. The clinical course of SLE is highly variable, marked by alternating periods of remission and disease flare-ups.
Definition
Autoimmunity: Immune system’s attack on self-antigens. Does not mean an autoimmune disease. Autoimmune disease is when autoimmunity causes signs and symptoms of a disease.
Antinuclear antibodies (ANA): Autoantibodies directed against nuclear antigens, hallmark of SLE.
Immune complex: Antigen-antibody aggregate that can deposit in tissues causing inflammation.
Lupus nephritis: Kidney inflammation due to immune complex deposition, a major complication of SLE.
Aetiology and Risk Factors
Multifactorial aetiology
- Genetics – common association with HLA-B8/DR3; ~10% have positive family history
- Complement deficiency C1q and C2
- TREX2 mutation
- Environment – UV radiation, cigarette smoking, infection, vitamin D deficiency
- Oestrogen
- Infection
- Certain drugs (hydralazine, procainamide, isoniazid — may cause drug-induced lupus)
Remember
Estrogen may enhance autoantibody production — a possible reason for female predominance.
Pathophysiology
Precipitating factors (factors that exacerbate and cause acute SLE reactions)
- Sun exposure (UV-B)
- Infections
- Stress
- Surgery
- Pregnancy
Pathophysiology
- Loss of immune tolerance → autoreactive T and B cells
- Production of autoantibodies (especially anti-dsDNA, anti-Smith)
- Formation of immune complexes → deposit in tissues
- Activation of complement and recruitment of inflammatory cells
- Tissue damage via type III hypersensitivity reaction
Think
Low complement (C3, C4) levels suggest ongoing immune complex consumption and active disease.
SLE can affect any organ system; key areas include:
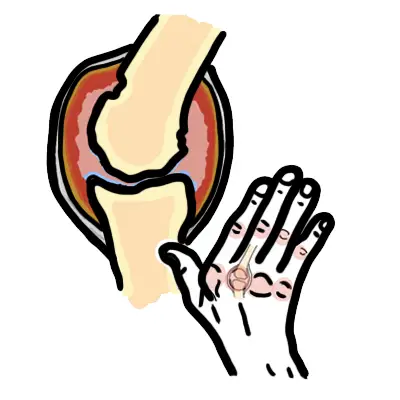
- Joints: Synovial tissue inflammation leads to arthralgia or arthritis
- Kidneys: Glomeruli are susceptible to immune complex deposition (e.g. Class III/IV lupus nephritis)
- Skin: UV-exposed areas show photosensitive rashes due to keratinocyte apoptosis
- CNS: Blood-brain barrier disruption may allow entry of neurotoxic antibodies
Clinical Manifestations
- Constitutional: Fatigue, fever, weight loss
- Skin:
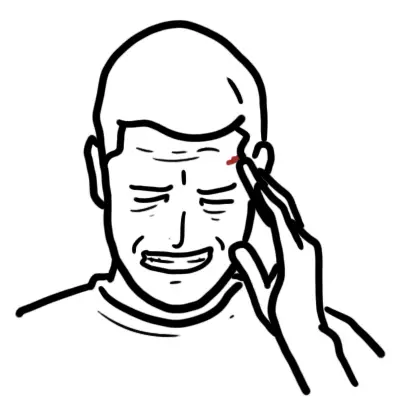
- Malar rash (butterfly rash sparing nasolabial folds)
- Discoid rash
- Photosensitivity
- Oral or nasal ulcers
- Joints: Non-erosive polyarthritis (often symmetric)
- Renal: Proteinuria, hematuria, cellular casts → Lupus nephritis
- Neurological: Seizures, psychosis, cognitive dysfunction
- Hematologic: Anemia, leukopenia, thrombocytopenia, lymphadenopathy
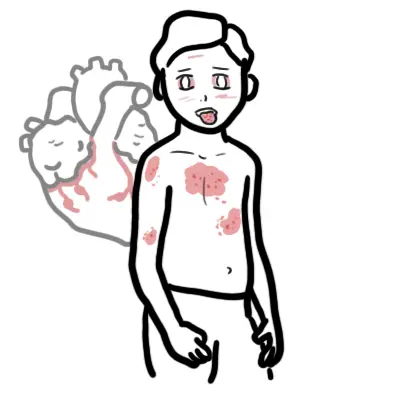
- Serosal: Pleuritis, pericarditis
- Cardiovascular: Raynaud’s phenomenon, livedo reticularis, Libman Sacks endocarditis
- Keratoconjunctivitis sicca
Remember
Malar rash, arthritis, and nephritis are among the most common presentations.
Think
In young females with multisystem symptoms, always consider SLE.
Diagnosis
- Screening: ANA (sensitive but not specific)
- Specific antibodies:
- Anti-dsDNA (specific and correlates with disease activity)
- Anti-Smith (highly specific but less sensitive)
- Other tests:
- CBC (cytopenias), urinalysis (proteinuria, casts), renal function
- Complement levels (C3/C4)
- ESR (often ↑), CRP usually normal unless serositis or infection
- Anti-phospholipid antibodies (linked to thrombosis)
- Biopsy: Renal biopsy for lupus nephritis classification or skin biopsy of cutaneous lupus
2023 ACR/EULAR SLE Classification Criteria
- Entry criterion: Positive ANA at a titer ≥1:80
- Additive weighted criteria include:
- Constitutional (fever), hematologic (leukopenia, thrombocytopenia)
- Mucocutaneous (malar rash, ulcers), renal, neurologic, serologic (autoantibodies, complement)
- Score ≥10 is classified as SLE
Differential diagnoses
| Condition | Key Differentiator |
| Rheumatoid Arthritis | Erosive arthritis, RF/anti-CCP + |
| Dermatomyositis | Muscle weakness, elevated CK, Gottron’s papules |
| Drug-induced lupus | Positive anti-histone, no nephritis or CNS. Typically arthritis and cutaneous manifestations |
| Mixed connective tissue disease | Overlap features + anti-U1 RNP |
Side note
SLE can overlap with other autoimmune diseases. For example SLE can occur with seropositive rheumatoid arthritis overlap typically referred to as Rupus.
Treatment
Non pharmacological
- Avoid overexposure to sunlight: sun protection and sunblock
- Smoking cessation
- Alcohol cessation
- Calcium and Vitamin D supplements
- Vaccinations
Pharmacological
- Mild disease (e.g. rash, arthritis):
- Hydroxychloroquine (mainstay)
- NSAIDs, short-term corticosteroids
- Moderate disease (serositis, cytopenias):
- Corticosteroids + immunosuppressants (azathioprine, methotrexate)
- Severe disease (nephritis, CNS):
- High-dose corticosteroids + cyclophosphamide or mycophenolate
- Biologics: Belimumab (anti-BLyS), Rituximab (off-label)
Remember
Hydroxychloroquine improves survival and reduces flares — baseline and annual eye exams are required after 5 years use.
Think
Always tailor immunosuppressants to organ system involvement and disease severity.
Complications and Prognosis
Complications
• Lupus nephritis → chronic kidney disease
• Neuropsychiatric lupus
• Increased infection risk due to immunosuppression
• Cardiovascular disease (accelerated atherosclerosis)
• Antiphospholipid syndrome (thrombosis, pregnancy loss)
• Osteoporosis (from corticosteroids)
Poor prognostic factors:
• Male gender
• Early onset nephritis
• Low complement, high anti-dsDNA
• African-American or Hispanic ethnicity
• Persistent cytopenias
Think
Most deaths are due to cardiovascular disease or infections in late disease.
Antiphospholipid Syndrome
Antiphospholipid Syndrome is an autoimmune prothrombotic disorder first described in the 1980s, characterized by arterial and venous thromboses and/or pregnancy morbidity in the presence of antiphospholipid antibodies—namely anticardiolipin (aCL), lupus anticoagulant (LAC), and anti-β2 glycoprotein I. APS can occur as a primary condition or secondary to another autoimmune disease, most commonly Systemic Lupus Erythematosus (SLE), with which it is associated in approximately 30% of cases.
A useful mnemonic for remembering the clinical features is CLOTS:
• Coagulation defect (arterial or venous thrombosis)
• Livedo reticularis (mottled skin appearance)
• Obstetric complications (especially recurrent miscarriage)
• Thrombocytopaenia (low platelet count)
• SLE association (present in ~30% of SLE cases)
Drug-Induced Lupus
Drug-Induced Lupus (DIL) is an autoimmune condition that mimics systemic lupus erythematosus (SLE) but is triggered by chronic exposure to certain medications. It typically presents with systemic features such as fever, arthralgia, myalgia, and serositis, but usually lacks the renal or central nervous system involvement seen in idiopathic SLE. The most commonly implicated drugs include hydralazine, procainamide, isoniazid, minocycline, and anti-TNF agents. Unlike SLE, DIL is more often associated with anti-histone antibodies and usually demonstrates a negative anti-dsDNA. Symptoms typically resolve within weeks to months after discontinuation of the offending agent, and long-term immunosuppressive therapy is rarely required.
Remember
Anti-histone antibodies are present in over 90% of DIL cases.
Remember
Renal and CNS involvement is rare.
Remember
Resolution occurs with drug withdrawal.
Think
In a patient with new-onset lupus-like symptoms and no prior autoimmune history, always review the medication list for potential triggers.
References
- Petri M, Orbai AM, Alarcón GS, et al. Derivation and validation of the SLICC classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677–2686.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis. 2019;78(9):1151–1159.
- Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110–2121.
- Fanouriakis A, Kostopoulou M, Cheema K, et al. 2023 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. 2023;82(1):48–61.
Discussion