

Cystic Fibrosis
Overview
Cystic fibrosis causes the dehydration of the mucus lumen causing thick sticky mucus to build up. This occurs mainly the lungs, causing productive cough and increasing rates in respiratory infections, and the pancreas causing pancreatic dysfunction. Cystic fibrosis occurs due to a mutation in the CFTR gene. Salty sweat is also another important characteristic of these patients.
Definition
CFTR: cystic fibrosis transmembrane conductance regulator; an epithelial chloride and bicarbonate channel.
Bronchiectasis: permanent abnormal dilatation and damage of the bronchi caused by chronic infection and inflammation.
Pancreatic exocrine insufficiency: inadequate pancreatic digestive-enzyme secretion causing maldigestion and malabsorption.
CFTR modulator: medicine that improves the production, processing or function of abnormal CFTR protein.
Meconium ileus: Neonatal bowel obstruction of the distal ileum due to abnormally thick and impacted meconium. Unlike in the meconium plug syndrome, the meconium is abnormal in consistency.
Aetiology & Risk Factors
Aetiology
- CF is caused by pathogenic variants involving both copies of the CFTR gene, located on chromosome 7.
- Inheritance is autosomal recessive:
- An affected person usually inherits one pathogenic variant from each parent.
- Parents are usually asymptomatic carriers.
- When both parents are carriers, each pregnancy carries:
- 25% probability of an affected child.
- 50% probability of a carrier child.
- 25% probability of a child without either familial variant.
- More than 2,000 CFTR variants have been described, although only a proportion are established as CF-causing.
- F508del is the most frequent pathogenic CFTR variant and primarily causes defective CFTR processing and trafficking.[1–4]
- Two parents carrying pathogenic CFTR variants.
- A sibling or close relative with CF.
- Known parental consanguinity.
- Previous pregnancy affected by CF.
- Ancestry from a population with a higher CFTR carrier frequency; however, CF can occur in any ethnic group.
CFTR has different physiological effects in different organs: it promotes airway and ductal secretion but facilitates salt reabsorption within sweat ducts.
Genetics
- CF is inherited in an autosomal recessive pattern.
- An affected person has pathogenic CFTR variants on both alleles.
- When both parents are carriers, each pregnancy has:
- 25% probability of an affected child
- 50% probability of a carrier child
- 25% probability of a child without either parental variant
- The F508del variant is the most common disease-causing CFTR variant in many populations.
- Genotype affects pancreatic function and eligibility for CFTR modulators, but does not precisely predict the severity of pulmonary disease in an individual.
Pathophysiology
Cystic Fibrosis is caused by mutated CTFR protein. The CFTR protein is found on the apical surface of many hollow tissues notably the lungs, pancreas, gastrointestinal tract and sweat glands.
The CTFR protein is a transporter for Cl-. A mutated CFTR protein can not transport Cl- properly and as a result this causes a disregulated ion distribution. Whereever Cl- exists Na+ will follow because of the electrochemical gradient. When Na+ move water tends to follow.
This basic principle is the cause of the signs and symptoms seen in Cystic Fibrosis
Chronological sequence in pulmonary disease
- Biallelic CFTR variants
- Pathogenic CFTR variants reduce the amount, stability or function of CFTR protein at the epithelial cell surface.
- Abnormal epithelial ion transport
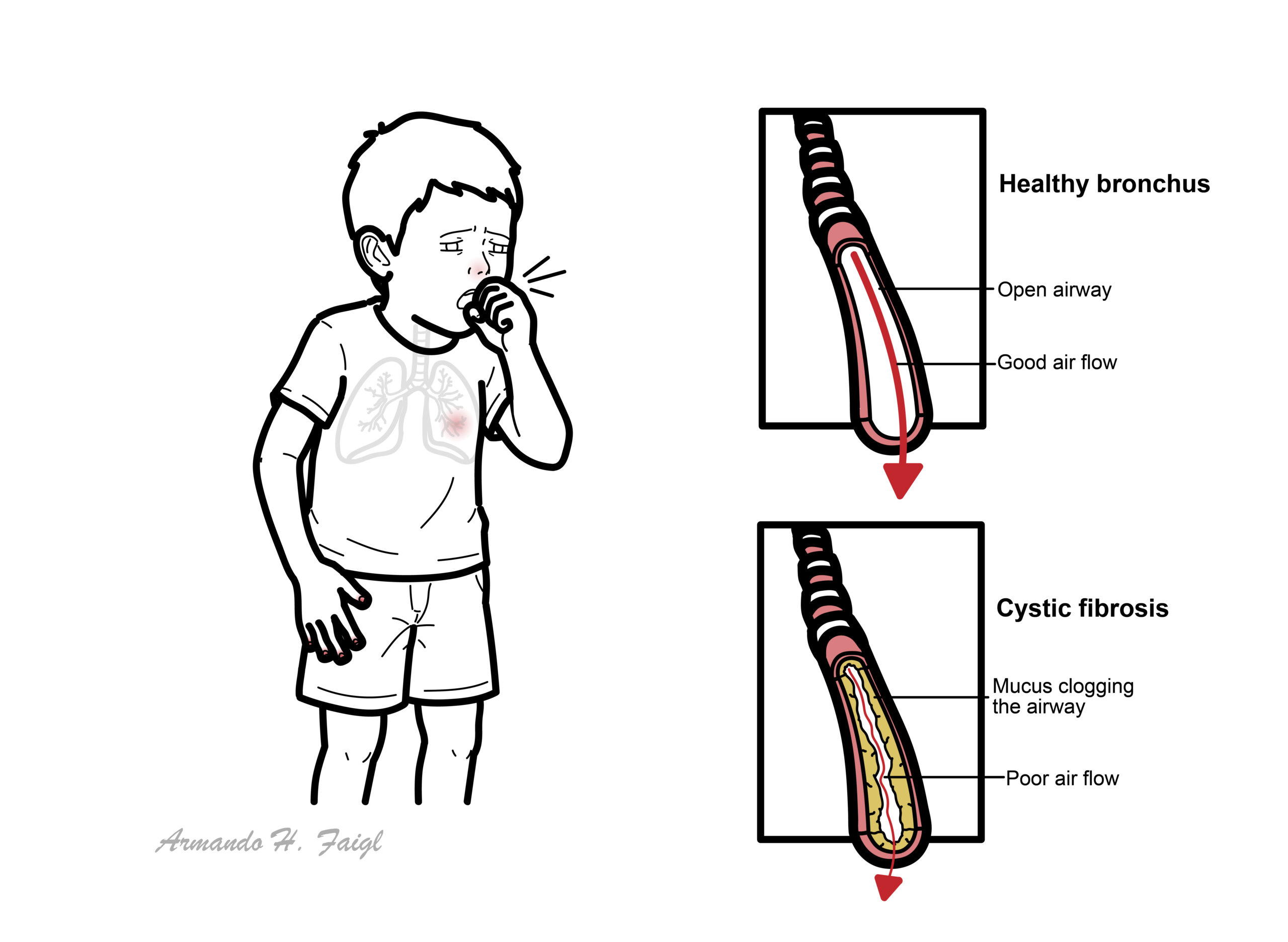
- Chloride and bicarbonate secretion is reduced.
- Sodium and water absorption may become excessive in the airway.
- Secretions become dehydrated, acidic and difficult to clear.
- Airway mucus obstruction
- Thick mucus adheres to airway surfaces.
- Ciliary movement and mucociliary clearance become ineffective.
- Small-airway obstruction and air trapping develop.
- Persistent airway infection
- Retained mucus provides a protected environment for bacterial growth.
- Early organisms commonly include S. aureus and Haemophilus influenzae.
- P. aeruginosa becomes increasingly important with age and may develop a biofilm-producing mucoid phenotype.
- Neutrophilic inflammation
- Persistent infection produces intense neutrophil recruitment.
- Neutrophil elastase, oxidants and extracellular DNA increase mucus viscosity and damage airway tissue.
- Structural lung disease
- Repeated obstruction, infection and inflammation cause bronchial wall destruction.
- Bronchiectasis, mucus plugging, fibrosis and progressive airflow limitation develop.
- Advanced pulmonary disease
- Ventilation–perfusion mismatch, hypoxaemia, hypercapnia, pulmonary hypertension and respiratory failure may occur.[1,2,5,6]
Lung damage results from a self-perpetuating cycle of obstruction, infection and inflammation.
Neutrophil-derived extracellular DNA is one reason CF sputum becomes extremely viscous.
Airway inflammation and structural damage may begin before prominent symptoms or abnormal conventional spirometry are present.
Extrapulmonary pathophysiology
- Pancreas:
- Intestine:
- Reduced luminal fluid and abnormally viscous contents cause meconium ileus in neonates and distal intestinal obstruction syndrome in older patients.
- Liver and biliary tract:
- Abnormally concentrated bile and focal duct obstruction cause inflammation, fibrosis, biliary cirrhosis and portal hypertension.
- Sweat glands:
- Failure to reabsorb sodium and chloride produces elevated sweat chloride and excessive salt loss.
- Reproductive tract:
- Abnormal fetal development and obstruction of the vas deferens cause congenital bilateral absence of the vas deferens in most males with classic CF.
- Thick cervical secretions and chronic illness may reduce female fertility.
- Paranasal sinuses:
- Mucus retention causes chronic rhinosinusitis and nasal polyposis.
Cystic Fibrosis is characterized by the triad of chronic obstructive pulmonary disease, pancreatic exocrine deficiency, and abnormally high sweat electrolyte concentrations.
Clinical Manifestation
The clinical presentation of cystic fibrosis defer in infants and children.
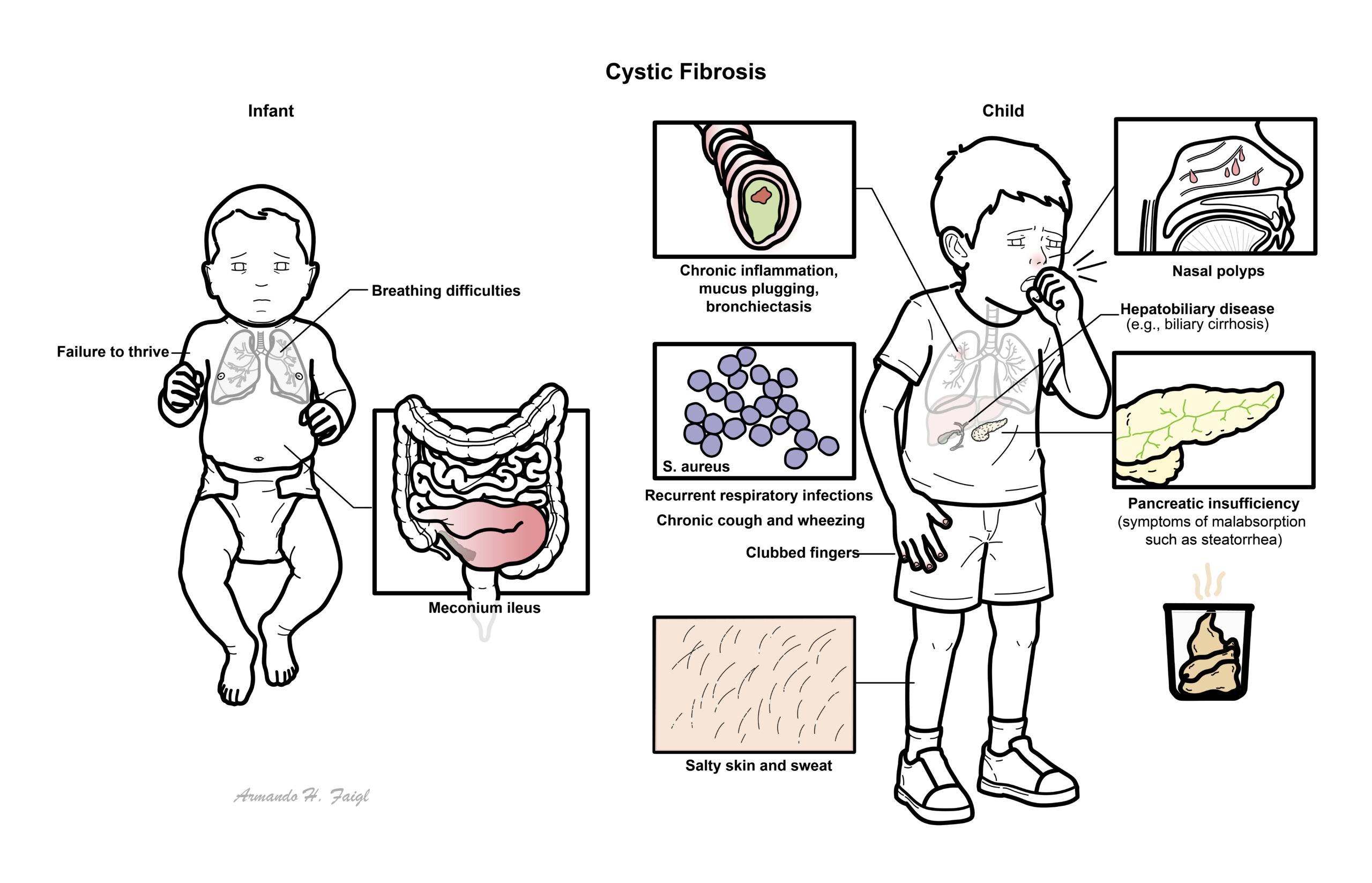
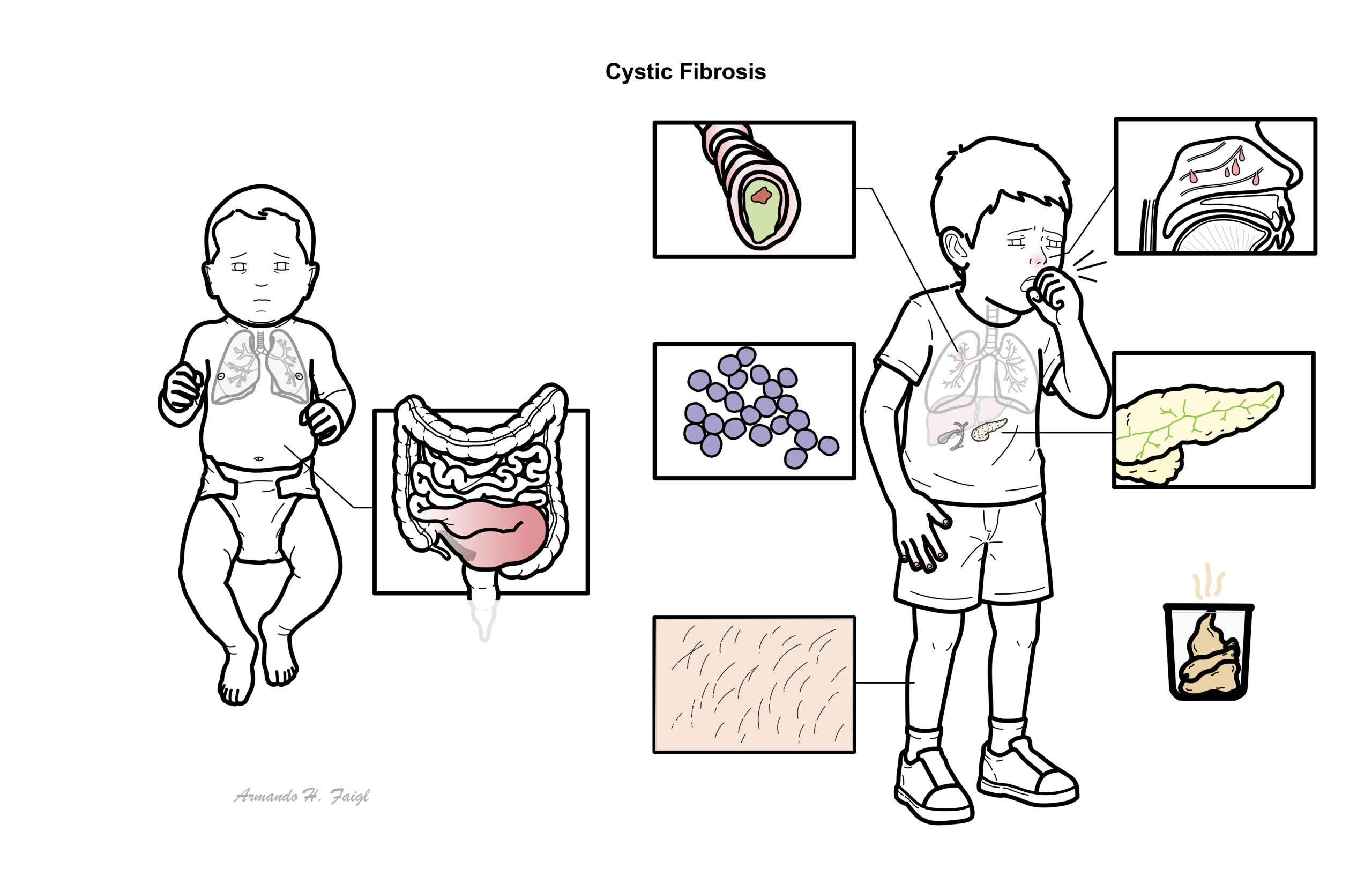
Infants
- Positive newborn screening result
- Meconium ileus
- Delayed passage of meconium
- Intestinal obstruction or volvulus
- Prolonged neonatal jaundice
- Failure to thrive despite an apparently adequate appetite.
- Rectal prolapse
Children – Mainly failure to thrive and recurrent upper respiratory tract infection
- Respiratory manifestations
- Persistent productive or wet cough.
- Recurrent lower respiratory tract infections.
- Wheeze, breathlessness and exercise limitation.
- Chronic rhinosinusitis.
- Nasal polyps, particularly in a child or young adult
- Gastrointestinal and nutritional manifestations
- Steatorrhoea.
- Abdominal distension and excessive flatulence.
- Pancreatic exocrine insufficiency.
- Distal intestinal obstruction syndrome.
- Gallstones.
- Endocrine and musculoskeletal manifestations
- CF-related diabetes.
- Delayed puberty.
- Fragility fractures.
- Kyphosis or postural abnormalities.
- Episodic CF-associated arthropathy.
- Reproductive manifestations
- Male infertility due to congenital bilateral absence of the vas deferens.
- Azoospermia despite generally preserved spermatogenesis.
Meconium ileus is strongly associated with CF and should trigger diagnostic testing.
Cystic Fibrosis usually presents in childhood as recurrent lung infections that become persistent and chronic.
Clinical examination
- Tachypnoea or increased work of breathing during an exacerbation.
- Hyperinflated chest.
- Coarse inspiratory crackles.
- Rhonchi or wheeze.
- Reduced air entry over areas of severe obstruction or collapse.
- Digital clubbing.
- Cyanosis in advanced disease.
- Signs of pulmonary hypertension or cor pulmonale, including a parasternal heave, loud pulmonary component of S2, elevated jugular venous pressure and peripheral oedema.
Diagnosis
- CF is diagnosed by:
- A compatible clinical presentation, positive newborn screen or family history; and
- Objective evidence of CFTR dysfunction.
- Newborn screening identifies infants requiring further investigation but is not itself diagnostic.[3,4]
- A diagnosis is supported by the identification of two CF-causing CFTR variants in trans.
| Current diagnostic criteria | ||
| Sweat chloride result | Interpretation | Recommended response |
|---|---|---|
| ≥60 mmol/L | Consistent with CF | Confirm on a separate occasion or by an independent diagnostic method |
| 30–59 mmol/L | Intermediate; CF remains possible | Repeat sweat testing, extended CFTR analysis and specialist functional testing where required |
| <30 mmol/L | CF is unlikely | Consider CF if the phenotype is strongly suggestive or two CF-causing variants are identified |
Newborn screening
- Heel prick
- Most programmes initially measure immunoreactive trypsinogen.
- Infants with a positive screen should undergo sweat testing when clinically stable and able to produce sufficient sweat.
Newborn screening can miss CF, particularly when uncommon CFTR variants are not included in the screening panel.
Heel prick is performed to screen for metabolic disease of the newborn inlcuding: galactosemia, cystic fibrosis, phenylketouria, sickle-cell disease and congenital hypothyroidism.
Baseline investigations following diagnosis
- Respiratory culture from sputum, cough swab or oropharyngeal sample.
- Spirometry in patients able to perform reliable testing.
- Chest imaging:
- Chest radiograph for broad assessment.
- Low-dose chest CT when detailed assessment of bronchiectasis, mucus plugging or air trapping is needed.
- Faecal elastase to assess pancreatic exocrine function.
- Fat-soluble vitamin concentrations.
- Full blood count, electrolytes and renal function.
- Liver biochemistry and platelet count.
- Abdominal ultrasound or elastography when hepatobiliary involvement is suspected or according to surveillance protocols.
- Oral glucose tolerance testing annually from approximately 10 years of age.
- Bone-density assessment according to age and risk factors.
Classification
| Classification | Typical features |
| Classic CF | Multisystem disease, pancreatic insufficiency, chronic sinopulmonary disease and clearly abnormal CFTR function |
| Non-classic or atypical CF | Later presentation, residual CFTR function, pancreatic sufficiency or limited organ involvement; still meets CF diagnostic criteria |
| CFTR-related disorder | Mono-organ or limited disorder associated with CFTR dysfunction but not meeting complete CF criteria |
| CRMS/CFSPID | Positive newborn screen with inconclusive diagnostic testing; applies primarily to asymptomatic screened infants |
| Functional CFTR variant classes | |||
| Class | Principal molecular abnormality | Example | Expected residual function |
| I | No functional protein synthesis | Nonsense or severe splice variants | Minimal or absent |
| II | Defective protein processing and trafficking | F508del | Minimal at cell surface |
| III | Defective channel regulation or gating | G551D | Protein present but channel rarely opens |
| IV | Reduced chloride conductance | R117H | Partial |
| V | Reduced synthesis of otherwise functional CFTR | Certain splice variants | Variable residual function |
| VI | Reduced stability at the cell surface | Several rare variants | Accelerated removal from membrane |
- Some variants produce more than one molecular defect.
- Functional class does not always reliably predict individual disease severity.
- CFTR-modulator eligibility should be based on the specific genotype, regulatory approval and evidence of responsiveness rather than class alone.[1,9]
Class II F508del is the most common CF-causing variant. Class III gating variants may respond markedly to a CFTR potentiator such as ivacaftor.
Treatment
General principles
- Management should be coordinated through a specialist multidisciplinary CF centre
- Annual influenza vaccination and routine age-appropriate vaccination.
- Avoid tobacco smoke and vaping.
- Infection-control precautions to reduce patient-to-patient transmission.
- Intranasal saline, topical corticosteroids and selected surgery for chronic rhinosinusitis.
- Vitamin D, calcium, weight-bearing exercise and bisphosphonates when indicated for bone disease.
- Psychological screening and treatment for anxiety, depression and treatment-related burden.
- Fertility counselling and assisted reproductive techniques when required.
- Genetic counselling for patients, siblings, partners and prospective parents.
CFTR-modulator therapy
- CFTR modulators address the underlying molecular defect.
- Main categories include:
- Potentiators: Increase the opening probability of CFTR channels already present at the cell surface, for example ivacaftor.
- Correctors: Improve CFTR folding, processing and trafficking, for example elexacaftor, tezacaftor and lumacaftor.
- Common regimens include:
- Ivacaftor for eligible gating and residual-function variants.
- Tezacaftor–ivacaftor for selected responsive variants.
- Elexacaftor–tezacaftor–ivacaftor for patients with at least one F508del variant or another approved responsive genotype.
Airway-clearance treatment
- Perform regular, usually daily, airway-clearance therapy.
- Increase frequency during respiratory exacerbations.
- Techniques include:
- Active cycle of breathing.
- Positive expiratory pressure.
- Oscillating positive expiratory pressure.
- Autogenic drainage.
- Percussion and postural drainage.
- High-frequency chest-wall oscillation.
- No single technique is universally superior; selection should account for age, preference, adherence and clinical response.
Mucoactive and inhaled therapies
- Dornase alfa:
- Recombinant human DNase.
- Cleaves extracellular DNA released by degenerating neutrophils.
- Reduces sputum viscosity and pulmonary exacerbations.
- Hypertonic saline:
- Draws water into the airway lumen.
- Improves mucus hydration and clearance.
- Inhaled mannitol:
- May be considered in selected older patients according to local availability.
- Bronchodilator:
- May be administered before hypertonic saline or inhaled antibiotics in patients with bronchial hyperresponsiveness.
- Inhaled antibiotics are generally administered after airway clearance to improve distal deposition.[2,5,6]
Antimicrobial management
- Obtain regular respiratory cultures even when the patient is clinically stable.
- First or newly detected P. aeruginosa:
- Commence eradication therapy promptly.
- A commonly recommended regimen is inhaled tobramycin for 28 days.
- Chronic P. aeruginosa infection:
- Suppressive inhaled antibiotics may include tobramycin, aztreonam or colistin according to local protocols.
- Regimens may be cycled or continuously alternated in selected patients.
- Long-term azithromycin:
- May reduce exacerbations and provide anti-inflammatory effects, particularly in patients with persistent P. aeruginosa.
- Nontuberculous mycobacterial infection should be considered before prolonged macrolide monotherapy.
- Pulmonary exacerbations:
- Increase airway clearance and optimise nutrition.
- Use oral, inhaled or intravenous antibiotics according to severity, previous cultures and susceptibility patterns.
- Severe exacerbations or resistant P. aeruginosa commonly require intravenous antipseudomonal therapy.
- Assess clinical recovery, weight and lung function after treatment.
- Routine long-term prophylactic antistaphylococcal therapy is not universally recommended.[5,6,10]
Pancreatic and nutritional management
- Assess pancreatic function using faecal elastase.
- Commence pancreatic enzyme replacement therapy in pancreatic-insufficient patients.
- Avoid excessive enzyme doses because of the risk of fibrosing colonopathy.
- Supplement vitamins A, D, E and K when deficient or in pancreatic-insufficient patients according to specialist guidance.
Gastrointestinal treatment
- Use polyethylene glycol and adequate hydration for constipation or distal intestinal obstruction syndrome.
CF-related diabetes
- Screen annually with a two-hour oral glucose tolerance test from approximately 10 years of age.
- HbA1c is not sufficiently sensitive as the sole screening test.
- Insulin is the preferred treatment for established CF-related diabetes.
Patients with CF are at risk of having recurrent URTI which can be caused by S. aureus, H. Influenzae, P. aerugenosa.
Complication & Prognosis
Respiratory
- Recurrent pulmonary exacerbations
- Bronchiectasis
- Chronic Pseudomonas infection
- Allergic bronchopulmonary aspergillosis
- Non-tuberculous mycobacterial disease
- Haemoptysis
- Pneumothorax
- Pulmonary hypertension
- Respiratory failure
Gastrointestinal and nutritional
- Pancreatic exocrine insufficiency
- Malnutrition and growth failure
- Fat-soluble vitamin deficiency
- Distal intestinal obstruction syndrome
- Pancreatitis
- Gallstones
- Rectal prolapse
- Increased colorectal-cancer risk in adults
Other complications
- CF-related diabetes
- CF-related liver disease and portal hypertension
- Osteopenia and osteoporosis
- Infertility
- Chronic sinusitis and nasal polyps
- Depression and anxiety
- Medication toxicity
- Renal impairment, particularly after repeated nephrotoxic antibiotics.
- CF remains a lifelong condition, but prognosis has improved considerably because of newborn screening, nutritional support, effective infection management, multidisciplinary care and CFTR modulators.
- Many people with CF now survive well into adulthood and pursue education, employment, relationships and parenthood.
- Progressive respiratory disease remains a major cause of morbidity and mortality.
- Lung transplantation may prolong survival in selected patients with advanced disease.
References
- Mall MA, Burgel PR, Castellani C, Davies JC, Salathe M, Taylor-Cousar JL. Cystic fibrosis. Nat Rev Dis Primers. 2024;10(1):53.
- Ong T, Ramsey BW. Cystic fibrosis: a review. JAMA. 2023;329(21):1859-1871.
- Farrell PM, White TB, Ren CL, et al. Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017;181S.e1.
- Castellani C, Duff AJA, Bell SC, et al. ECFS best practice guidelines: the 2018 revision. J Cyst Fibros. 2018;17(2):153-178.
- Southern KW, Addy C, Bell SC, et al. Standards for the care of people with cystic fibrosis: establishing and maintaining health. J Cyst Fibros. 2024;23(1):12-28.
- Burgel PR, Southern KW, Addy C, et al. Standards for the care of people with cystic fibrosis: recognising and addressing CF health issues. J Cyst Fibros. 2024;23(2):187-202.
- Wilschanski M, Munck A, Carrion E, et al. ESPEN-ESPGHAN-ECFS guideline on nutrition care for cystic fibrosis. Clin Nutr. 2024;43(2):413-445.
- Sellers ZM, Assis DN, Paranjape SM, et al. Cystic fibrosis screening, evaluation, and management of hepatobiliary disease consensus recommendations. Hepatology. 2024;79(5).
- Taylor-Cousar JL, Robinson PD, Shteinberg M, Downey DG. CFTR modulator therapy: transforming the landscape of clinical care in cystic fibrosis. Lancet. 2023;402(10408):1171-1184.
- Cogen JD, Quon BS. Update on the diagnosis and management of cystic fibrosis pulmonary exacerbations. J Cyst Fibros. 2024;23(4):603-611.
- Ruseckaite R, Salimi F, Earnest A, et al. Survival of people with cystic fibrosis in Australia. Sci Rep. 2022;12:19748.
- Australian Cystic Fibrosis Data Registry. Australian Cystic Fibrosis Data Registry Annual Report 2023. Melbourne: Monash University and Cystic Fibrosis Australia; 2024.







Members only discussions coming soon…