

Diabetes Insipidus
56 year old lady recurrent presentations to your practice for polyuria and increased thirst. Random serum glucose and urinary glucose have returned normal. Patient has otherwise been well, complains of no dysuria, haematuria, fevers or night sweats.
Overview
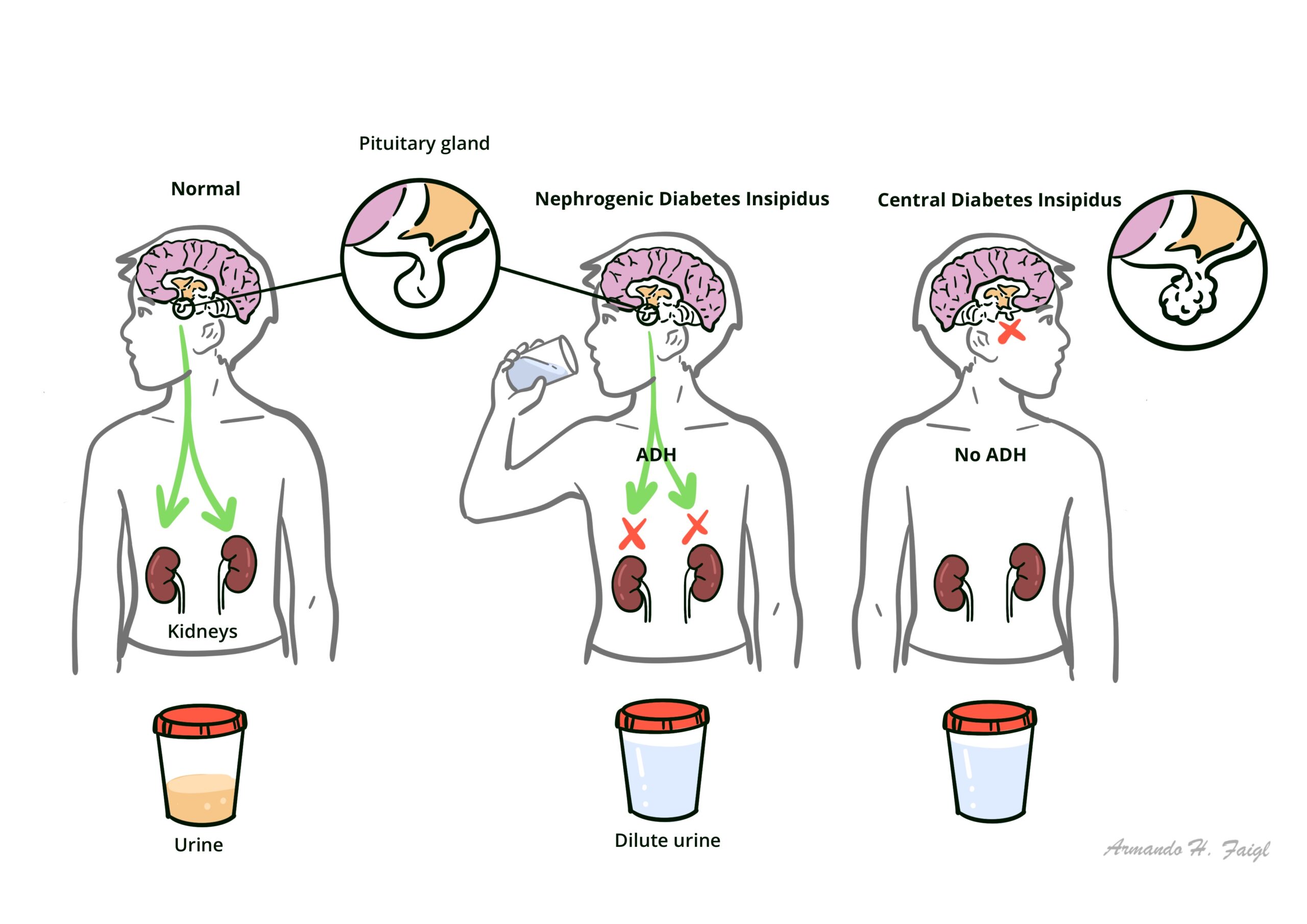
Diabetes Insipidus (DI) is associated with inadequate arginine vasopressin (known as antidiuretic hormone) secretion or renal response to arginine vasopressin, resulting in hypotonic polyuria and a compensatory/underlying polydipsia. There are two main types of diabetes insipidus, central DI and nephrogenic DI
Triad Polyuria, dilute urine, and increased thirst are characteristic of DI.
Definition
Diabetes Insipidus: Disorder resulting from deficiency of ADH or its action
Diabetes Mellitus: A group of metabolic diseases in which there are high blood sugar levels over a prolonged period.
ADH: Antidiuretic hormone also known as arginine vasopressin
SIADH: inappropriate secretion of ADH (opposite condition to diabetes insipidus)
Desmopressin: artificially acquired ADH (for treatment for those with ADH deficiency/unresponsiveness).
Physiology of ADH
Antidiuretic hormone is initially produced by the hypothalamus and then transported to the posterior pituitary gland via the pituitary stalk for storage.
When osmoreceptors sense hyperosmolarity in the blood it stimulates the posterior pituitary gland to release ADH into systemic circulation.
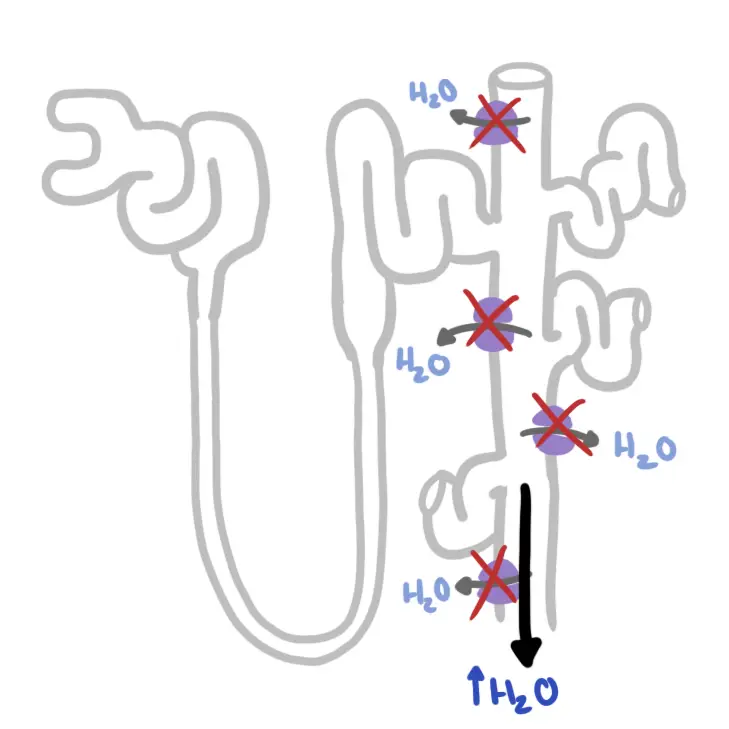
ADH acts on the kidneys and activates the arginine vasopressin 2 (AVP2) receptors of the renal collecting duct which increases the generation of aquaporin 2. Aquaporin 2 channels increases water retention
As a result, there is a net increase in water reabsorption in the collecting duct, leading to appropriate reservation of water and concentrating urine.
To little ADH or lack of response of the kidney to ADH means less water retention and more water output resulting in polyuria.
To little ADH or lack of response of the kidney to ADH means less water retention and more water output resulting in polyuria.
Aetiology & Risk Factors
Aetiology
Central DI (also known as neurogenic DI) caused by insufficient synthesis or release of ADH from the central nervous system.
Nephrogenic DI caused by ineffective response to ADH in the kidneys, such as defective ADH receptors caused by genetic defects.
Dipsogenic DI (also known as primary polydipsia) results from excessive fluid intake practiced over an extended period. Sometimes not classified as a true diabetes insipidus.
Gestational DI During pregnancy, vasopressins are more readily metabolised peripherally by placenta hormones. Hence it can provoke transient “central DI” in some patients. Commonly, this condition resolves spontaneously upon delivery.
Central DI
- Existing pituitary stalk lesions (pituitary adenoma)
- Traumatic head injury
- Congenital pituitary malformations
- Wolfram’s syndrome
- Autoimmune disorders (Hashimoto’s thyroiditis)
Wolfram syndrome is a genetic autosomal recessive disorder with the Tetrad: diabetes insipidus, diabetes mellitus, optic atrophy and deafness.
Nephrogenic DI
- Lithium therapy
- Chronic kidney disease
- Chronic hypercalcaemia
- Chronic hypokalaemia
- AVP receptor pathway mutations
Pathophysiology
Central Diabetes Insipidus
Traumatic or pathological damages affecting the hypothalamus or posterior pituitary gland causes cell death in hormone secreting cells in those areas, thus affecting the normal secretion and release of ADH. Without appropriate stimulation of ADH in the kidneys, renal collecting ducts lost its ability to perform adequate water reabsorption essential for volume maintenance of the body, resulting in a diuretic phenomenon.
Nephrogenic Diabetes Insipidus
Nephrogenic DI is caused by defective ADH receptors in the kidneys. Normally, two receptors AVPR1 and AVPR2 responds to increasing levels of ADH in the systemic circulation. AVPR1 is responsible for vasoconstriction and prostaglandin release, whereas the AVPR2 receptors mediated the antidiuretic response as well as certain coagulation factors (factor VIII and von Willebrand’s factor), hence unresponsive AVPR2 receptors in nephrogenic DI causes diuretic effects as well as mild coagulation defects.
Clinical Manifestations
General
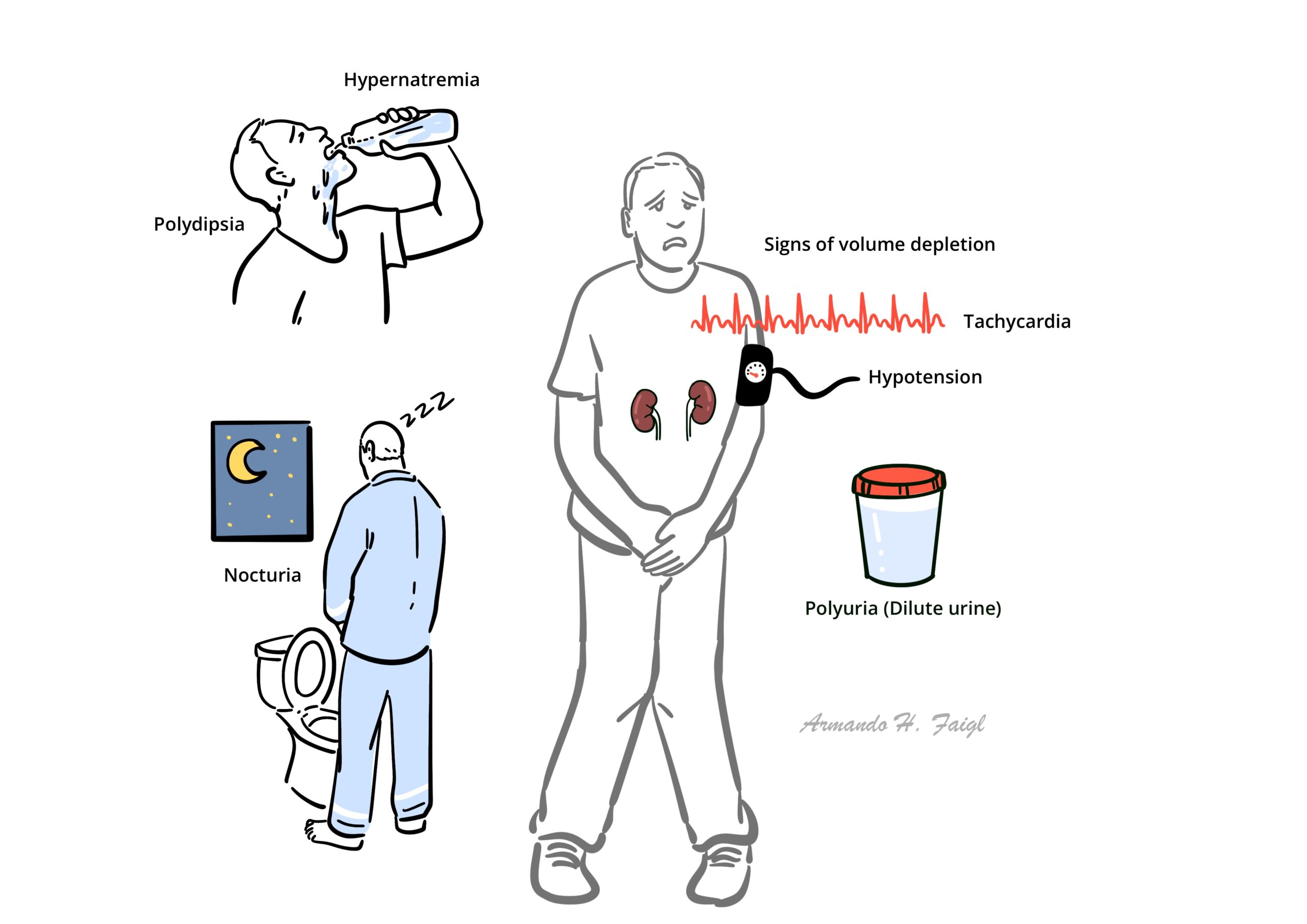
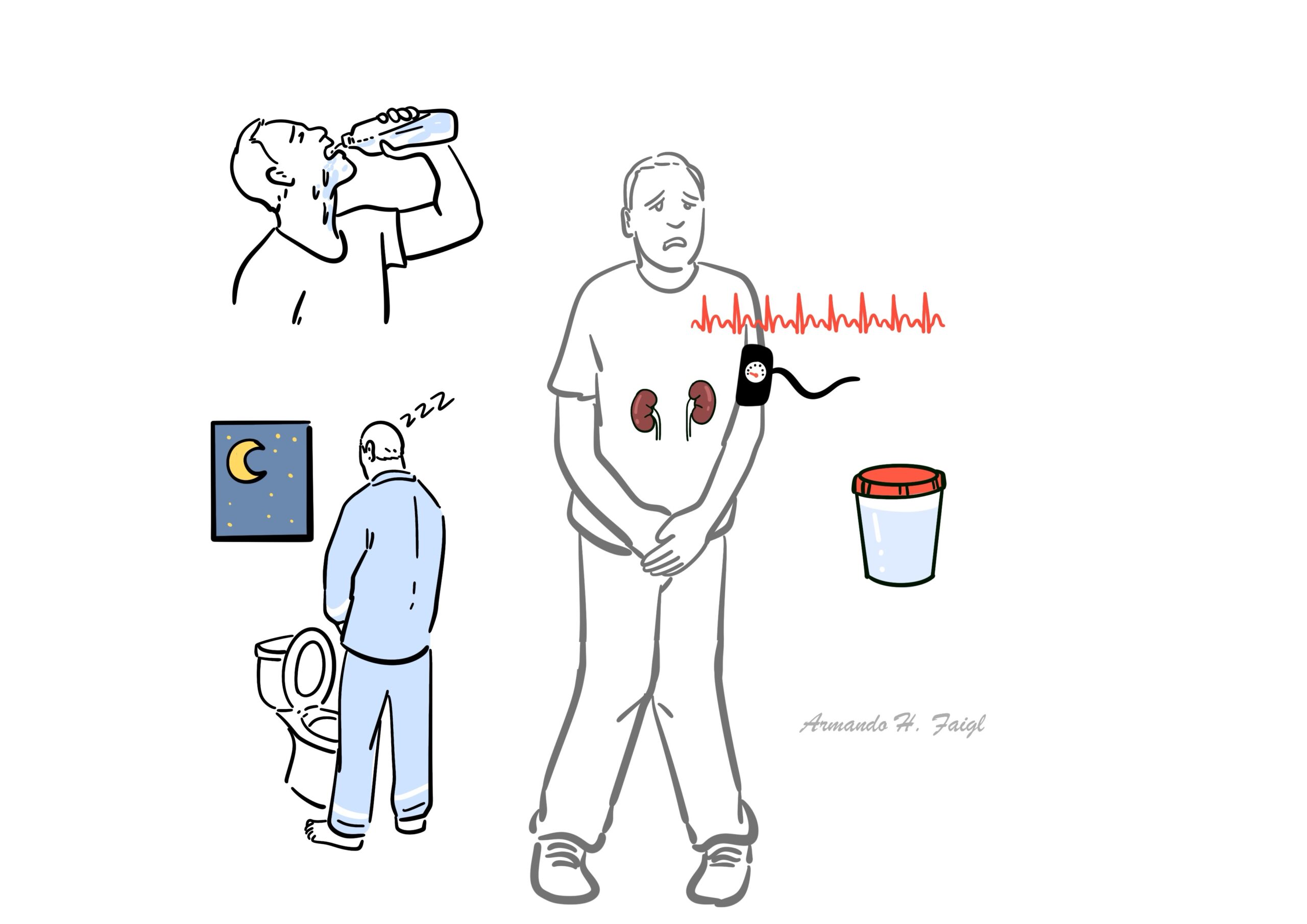
- Polydipsia
- Polyuria
- Nocturia
- Signs of hypernatraemia
- Signs of volume depletion (Tachycardia, hypotension)
Patients with traumatic brain injury may develop a 3-phase response:
- Polyuric phase (decrease in ADH, immediate increase in urine output + decrease urine osmolality, 4-5 days)
- Antidiuretic phase (axonal necrosis of ADH secreting neurons, uncontrolled ADH release with the reduction in urinary output and increase urine osmolality, 5-6 days)
- Permanent central DI (neuron death, with cessation of ADH production and depletion of ADH reserve)
Differential Diagnosis – polyuria and polydipsia
- Diabetes Mellitus Type I
- Diabetes Mellitus Type II
- Benign prostate hyperplasia
- Excessive fluid intake
- Pituitary adenoma
- Craniopharyngioma
- Psychogenic polydipsia
- Hyperaldosteronism
- Medications (Diuretics overdose)
- Hypercalcaemia
- Hyperosmolar hyperglycaemic state (HHS)
- Urinary tract obstructions (prostatic hypertrophy, osmotic diuresis)
Investigations
- 24 hours urine collection (volume) – typically 3-20 L of urine per day
- Urine osmolality – low results <300 mmol/kg
- Serum osmolality – normal or elevated
- EUC – elevated serum sodium
Elevated serum sodium with hypotonic urine strongly suggest DI, serial measures are necessary.
- Urinalysis – exclude other causes such as Diabetes mellitus (DM)
- Serum glucose – determine coexisting DM
Water deprivation test/dehydration test
- AVP (desmopressin stimulation test) – differentiating CDI or NDI – responds to AVP = central (respond by reduction in urine output and increase urine osmolality of >50%
- Genetic studies – considered more in paediatric patients
Treatment
Hypernatraemia management (regular complication of Diabetes insipidus)
- IV hypotonic fluids (5% dextrose and 0.45% sodium chloride)
- Frequent monitoring of electrolytes
Central Diabetes insipidus
- Desmopressin
- Oral or IV fluid replacement (only in acute settings)
Nephrogenic diabetes insipidus
- Maintenance of adequate PO fluid
- High dose desmopressin
- Sodium restriction
- Hydrochlorothiazide
- Other pharmacological therapy
- Treat underlying cause
- Stop or review medication dosage (diuretics)
- Treat uncontrolled DM
- Correct chronic hypercalcaemia or hypokalaemia
Complications & Prognosis
- Hypernaturaemia
- Growth retardation
- Hydronephrosis
For patients with significant underlying aetiology, DI typically resolves following the resolution of the underlying condition (e.g. correction of hypercalcaemia may result in resolving the presenting DI). With one exception that for lithium therapy induced DI, the condition is usually irreversible.
For patients with irreversible DI, lifelong follow up with monitoring is necessary, with regular assessments of their fluid status, electrolyte and renal functions. They are generally stable with good control by desmopressin, and if indicated, pituitary hormone replacement.
References
- Up to Date
- Best practice
- Grey’s anatomy







Members only discussions coming soon…