

Myotonic Dystrophy

Overview
Autosomal dominant disease characterised by trinucleotide repeat of CTG. Also known as Dystrophica Myotoncia the disease is causes an increase in muscle tone (Myotonia) with associated degeneration and shrinkage of muscle fibres (Dystrophy).
Definition
Myotonia: Delayed relaxation of skeletal muscle following voluntary contraction or mechanical stimulation (e.g., persistent grip after a handshake or a persistent dimple on percussion of the thenar eminence).
Anticipation: The clinical phenomenon where a genetic disease increases in severity and presents at an earlier age in successive generations, driven by progressive expansion of unstable nucleotide repeats during gametogenesis.
RNA Toxic Gain-of-Function: The primary pathogenic mechanism wherein mutant transcripts containing expanded nucleotide repeats aggregate into nuclear foci, sequestering essential RNA-binding proteins.
Spliceopathy: Abnormal alternative splicing of downstream target precursor mRNAs across multiple tissues, caused by functional depletion of splicing regulation factors like MBNL1.
Classification
Myotonic dystrophy is divided into two primary genetic subtypes, with DM1 further subdivided by age of onset.
1. Myotonic Dystrophy Type 1 (DM1 / Steinert Disease)
- Prevalence: Accounts for ~98% of all myotonic dystrophy cases.
- Genetics: CTG repeat expansion in the 3′ untranslated region (UTR) of the DMPK gene on chromosome 19.
2. Myotonic Dystrophy Type 2 (DM2 / Proximal Myotonic Myopathy – PROMM)
- Genetics: CCTG repeat expansion in intron 1 of the CNBP gene on chromosome 3.
- Features: Proximal muscle weakness (rather than distal), milder clinical course, no congenital form, and minimal genetic anticipation.
DM1 causes distal muscle weakness (facial, neck flexors, foot drop), whereas DM2 causes proximal muscle weakness (hip flexors, shoulders).
Aetiology & Risk Factors
1. Aetiology & Genetics
- DM1: Autosomal dominant inheritance of unstable CTG repeats in DMPK.
- Normal: 5–37 repeats.
- Premutation: 38–49 repeats.
- Symptomatic Disease: > 50 repeats (can expand to thousands).
- DM2: Autosomal dominant inheritance of unstable CCTG repeats in CNBP.
- Normal: < 30 repeats.
- Symptomatic Disease: 75 to > 11,000 repeats.
2. Risk Factors & Transmission Dynamics
- Family History: Positive family history of neuromuscular disease, unexplained early-onset cataracts, or early cardiac pacemakers.
- Maternal Transmission Risk: Congenital DM1 occurs almost exclusively when the expanded allele is inherited from the mother, due to massive intergenerational CTG expansion during maternal oogenesis.
Congenital DM1 is almost exclusively inherited from the mother due to massive CTG repeat expansion during oogenesis.
Pathophysiology
- Genetic mutations -> abnormal mRNA
- abnormal mRNA like these do not leave the nucleus
- The abnormal mRNA can cause these hairpin structures which disrupt existing proteins in the nucleus
- The abnormal mRNA causes existing proteins to lose function or gain function:
- Loss of function of the MBNL proteins
- Gain of function of CUGBP proteins
- These proteins are normally involved in splicing, mRNA transport, stability and decay
- These protein dysfunction are thought to be the pathological hallmark of Myotonic Dystrophy resulting in:
- Disruption of alternative splicing
- Disruption mRNA transport
- Disruption of mRNA decay
- Abnormal protein production and function causing complications of Myotonic dystrophy.
Clinical Manifestation
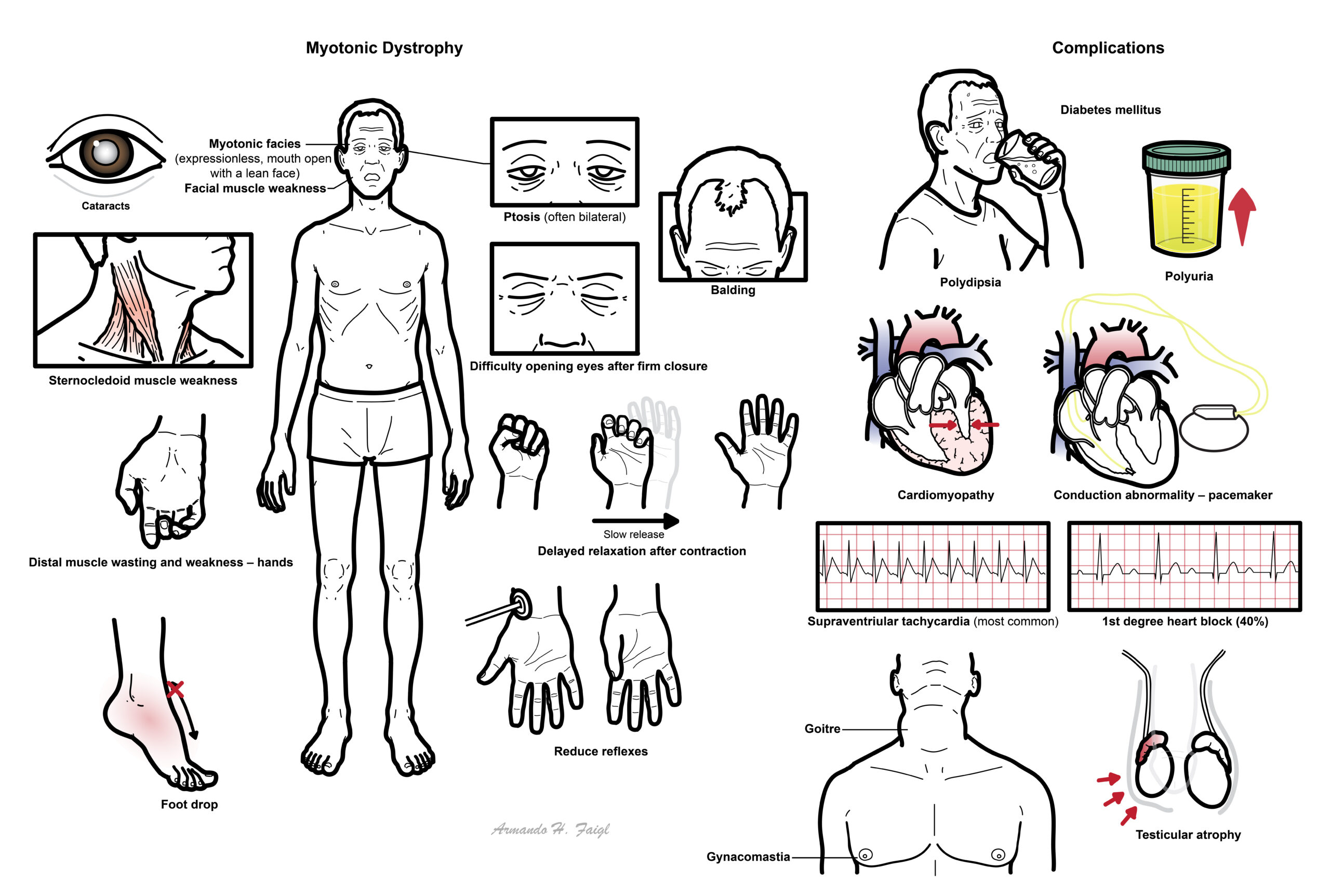
Myotonia is defines as continued contraction of the muscle after voluntary contraction ceases.
- Delayed relaxation after contraction – hand shake slow to release
- Myotonic facies (expressionless, mouth open with a lean face)
- Facial muscle weakness
- Sternocledoid muscle weakness
- Difficulty opening eyes after firm closure
- Ptosis (often bilateral)
- Cataracts
- Balding
- Distal muscle wasting and weakness – hands
- Reduce reflexes
- Foot drop
Myotonic dystrophy type 2 has all the same features of the classic myotonic dystrophy with the difference being in proximal muscle wasting/weakness rather than distal.
Complications
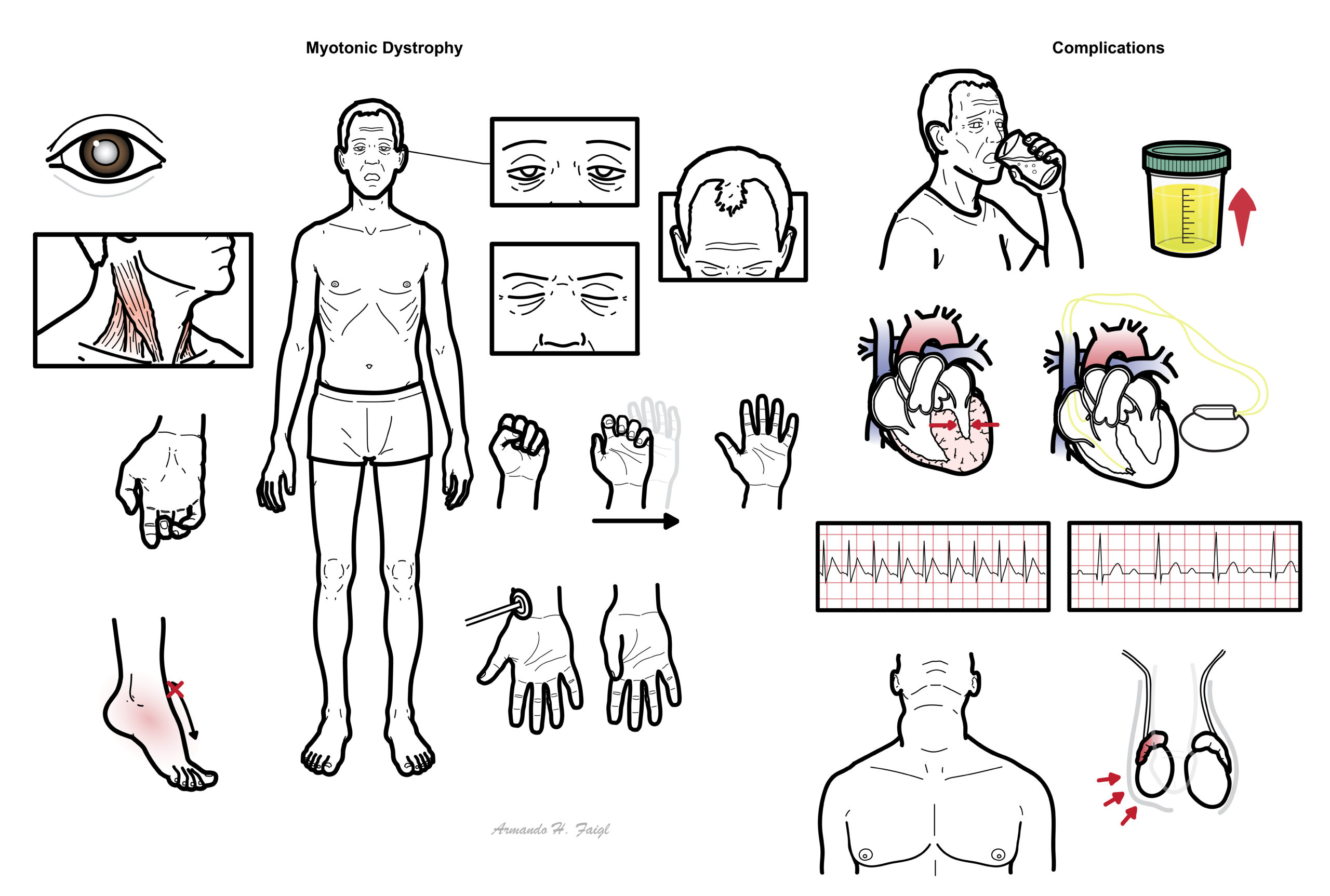
- Diabetes – polyuria, polydipsia
- Cardiomyopathy – features of heart failure
- Conduction abnormality – pacemaker
- Supraventriular tachycardia (most common)
- 1st degree heart block (40%)
- Hypogonadism – gynacomastia and testicular atrophy
- Goitre
What other disorders involve trinucleotide repeat?
Diagnosis
Diagnosis is by Molecular Genetic Testing (Gold Standard)
- Targeted Mutation Analysis: PCR and Southern Blot analysis of peripheral blood leukocytes to measure CTG (DM1) or CCTG (DM2) repeat numbers. Confirms diagnosis without needing a muscle biopsy.
- Electromyography (EMG)
- Myotonic Discharges: High-frequency repetitive muscle fiber action potentials that wax and wane in amplitude and frequency.
- Auditory Finding: Produces a pathognomonic “dive-bomber” sound on the loudspeaker.
Other investigations are mainly to look for complications of Myotonic Dystrophy
- Ophthalmology review
- Electrocardiogram
- 24 holter monitor
- Echocardiogram
- Sleep study
- Pulmonary function test
- HbA1c
- TFT
Treatment
- Medications that reduce sustained myotonia
- sodium channel blockers such as mexiletine
- tricyclic antidepressants
- benzodiazepines
- calcium antagonists
Treatment again targets complications of Myotonic Dystrophy
- Pacemaker, ICD
- CPAP machine
- Incentive spirometry
- Insulin
- Thyroxine
- Speech pathology for dysphagia
- Physical and occupational therapy is recommended for strengthening weakened muscles, evaluation for orthotics, and durable medical equipment needs.
Always obtain a 12-lead ECG before starting Mexiletine for myotonia, as it is strictly contraindicated in patients with underlying cardiac conduction blocks.
Complications
Endocrine complications
- Type II diabetes melitius
- Hypogonadism
- Nodular thyroid enlargement
Cardiovascular complications
- Resting ECG changes – everything prolonged (PR, QRS, QT)
- Arrhythmia
- Supraventricular tachycardia (most common)
- VT
- Conduction blocks
- 1st (common)
- 2nd
- complete
- Mitral valve prolapse
- Cardiomyopathy
Patients with Myotonic Dystrophy are at extreme risk during anesthesia due to exaggerated sensitivity to muscle relaxants and opioids, high risk of aspiration, and severe baseline cardiac/respiratory fragility. Succinylcholine can trigger life-threatening generalized myotonia and must be avoided
Gastrointestinal complications
- Dysphagia
- Reflux
- Delayed gastric emptying
- Malabsorption
- Bacterial overgrowth
- Megacolon
Respiratory complications
- Hypoventilation – poor sleep
- Respiratory failure follow anaesthesia
- Recurrent pneumonia
References
- Thornton CA. Myotonic dystrophy. Neurol Clin. 2014;32(3):705-719. doi:10.1016/j.ncl.2014.04.011
- Meola G, Cardani R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathogenesis. Acta Myol. 2015;34(1):6-20.
- Heatwole C, Bode R, Johnson NE, et al. Patient-reported impact of myotonic dystrophy type 1: establish the DM1-HI. Muscle Nerve. 2012;46(3):324-333. doi:10.1002/mus.23301







Members only discussions coming soon…