Overview
X-linked agammaglobulinaemia (XLA) is a rare primary immunodeficiency caused by mutations in the Bruton’s tyrosine kinase (BTK) gene, leading to failure of B-cell maturation. It results in profound hypogammaglobulinaemia and susceptibility to recurrent bacterial infections, particularly with encapsulated organisms. It usually presents after 6 months of age (once maternal IgG wanes). Prevalence is ~1 in 200,000 live male births.
Triad “Bruton’s = Boys, B-cells, Bacteria.”
Definition
BTK (Bruton’s tyrosine kinase): Enzyme critical for B-cell maturation.
Hypogammaglobulinaemia: Low levels of immunoglobulins (IgG, IgA, IgM).
Encapsulated bacteria: Bacteria such as S. pneumoniae, H. influenzae that require opsonising antibodies for clearance.
Opsonisation: Process where antibodies coat bacteria to promote phagocytosis.
Anatomy & Physiology
- Normal B-cell development: Pro-B cell → pre-B cell (BTK required) → immature B cell → plasma cell producing antibodies.
- In XLA, mutation in BTK halts differentiation at the pre-B stage, leading to:
- Absent circulating B cells.
- Absent plasma cells.
- Absent immunoglobulins (IgG, IgA, IgM, IgE).
Remember
“XLA = no B cells, no antibodies.”
Aetiology & Risk Factors
Aetiology
- Genetics: Mutations in BTK gene (Xq21.3–Xq22).
- Inheritance: X-linked recessive → almost exclusively affects boys.
Risk Factors
- Family history of affected males.
- Carrier mothers (asymptomatic).
Pathophysiology
- BTK mutation → defective pre-B to mature B-cell transition.
- Absent mature B cells and plasma cells.
- Profound hypogammaglobulinaemia.
- No antibody-mediated opsonisation → impaired clearance of encapsulated bacteria.
- Recurrent bacterial infections from ~6 months of age (loss of maternal IgG).
Think
Timing is key – healthy until 6 months, then recurrent infections.
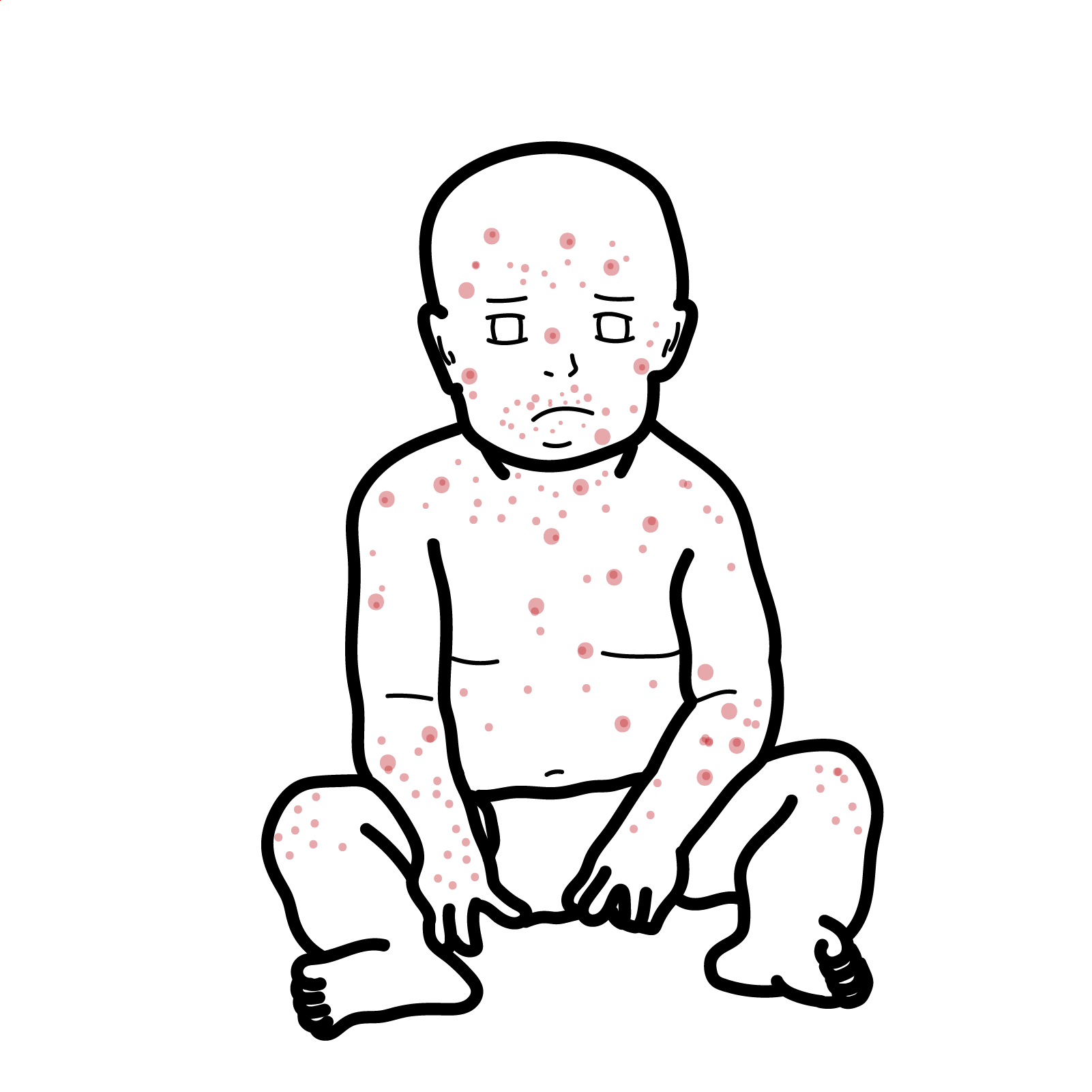
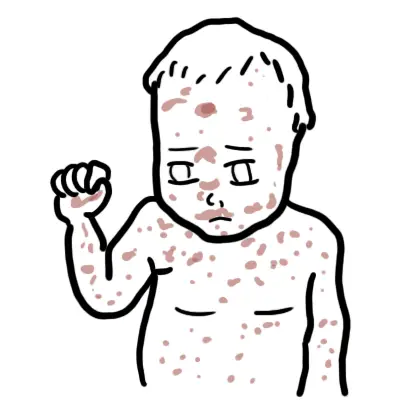
Clinical Manifestations
- Onset after 6 months of age.
- Recurrent bacterial infections:
- Otitis media, sinusitis, pneumonia.
- Septicaemia, meningitis.
- Organisms: Streptococcus pneumoniae, Haemophilus influenzae, Enteroviruses (esp. polio, echovirus).
- GI infections: Chronic diarrhoea from Giardia lamblia.
- Absent lymphoid tissue: Very small or absent tonsils, adenoids, lymph nodes.
- Chronic lung disease: Bronchiectasis if untreated.
Remember
“Boy with recurrent bacterial infections + absent tonsils” = XLA.
Diagnosis
- CBC: Normal lymphocyte count.
- Immunoglobulins: Profound ↓ IgG, IgA, IgM.
- Flow cytometry: Absent/very low CD19+ B cells.
- Genetic testing: BTK mutation.
- Physical exam: Absent tonsils/lymph nodes.
Differential Diagnosis
| Condition | Differentiating Feature |
| CVID | Later onset, some B cells present, not complete absence |
| Selective IgA deficiency | Only IgA absent, often asymptomatic |
| SCID | Severe infections earlier (<3 months), T- and B-cell defects |
| Secondary hypogammaglobulinaemia | Drug-induced (rituximab, steroids), protein loss |
Treatment
- Lifelong IVIG or SCIG replacement therapy.
- Prompt antibiotic therapy for infections.
- Prophylactic antibiotics may be used.
- Avoid live vaccines (esp. oral polio)
- Aggressive management of chronic lung disease (bronchiectasis).
Think
Treatment is supportive (antibodies supplied), no curative therapy currently.
Complications & Prognosis
- Recurrent severe infections → bronchiectasis, chronic lung disease.
- Enteroviral meningoencephalitis.
- Arthritis (from Mycoplasma or Ureaplasma).
- Prognosis: excellent with immunoglobulin replacement and infection prevention.
Remember
Without treatment, most die in childhood; with Ig replacement, can live near-normal lifespan.
References
- Bruton OC. Agammaglobulinemia. Pediatrics. 1952;9(6):722–728.
- Conley ME, Dobbs AK, Quintana AM, et al. Genetic basis of X-linked agammaglobulinemia. N Engl J Med. 2009;360(7):676–81.
- Winkelstein JA, Marino MC, Lederman HM, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore). 2006;85(4):193–202.
- Ochs HD, Smith CIE. X-linked agammaglobulinemia: a model primary immunodeficiency. J Allergy Clin Immunol. 1996;98(4):687–99.
- Picard C, Al-Herz W, Bousfiha A, et al. Primary immunodeficiency diseases: 2015 IUIS classification. J Clin Immunol. 2015;35(8):696–726.







Discussion