

Nephritic Syndrome

Overview
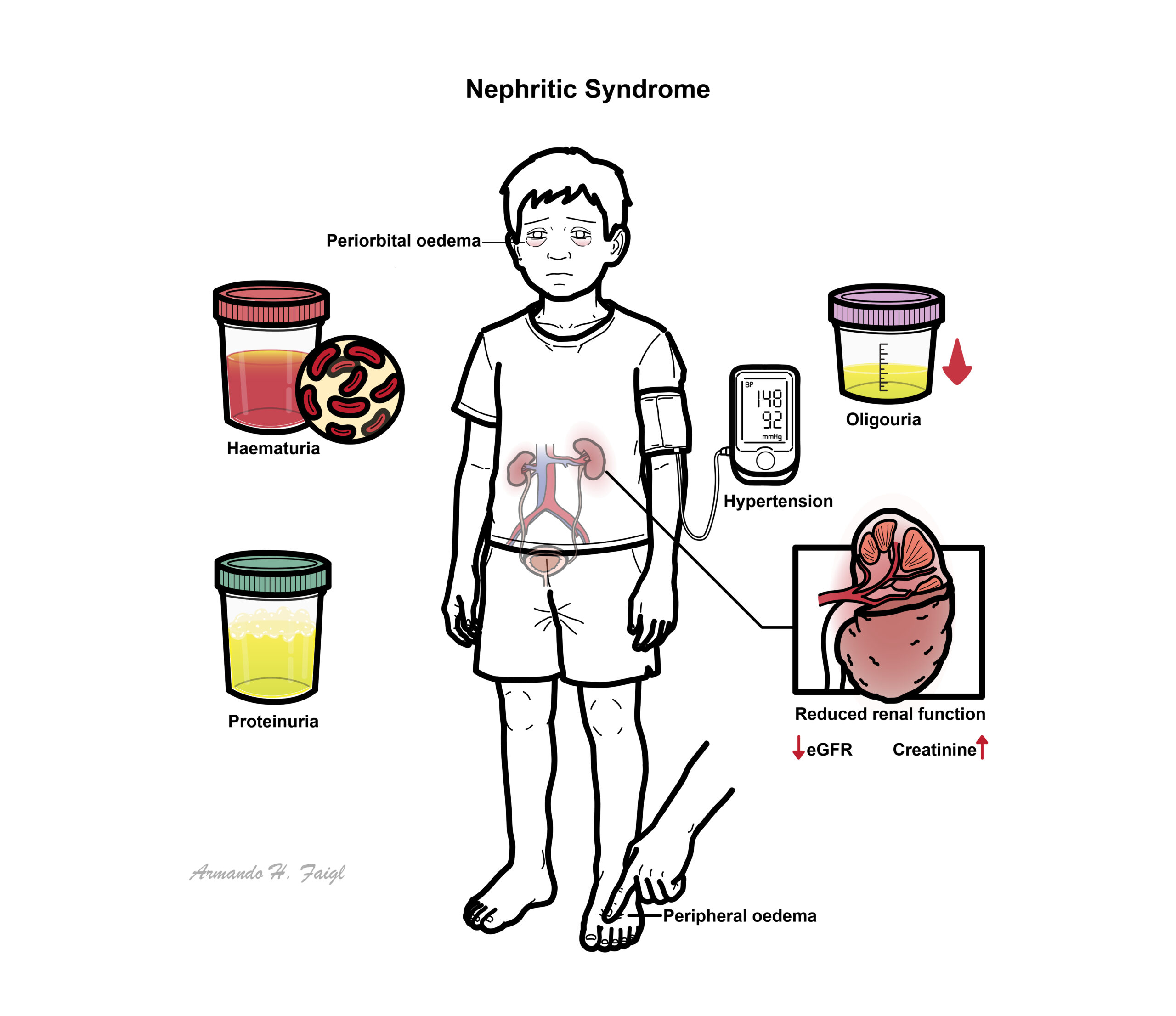
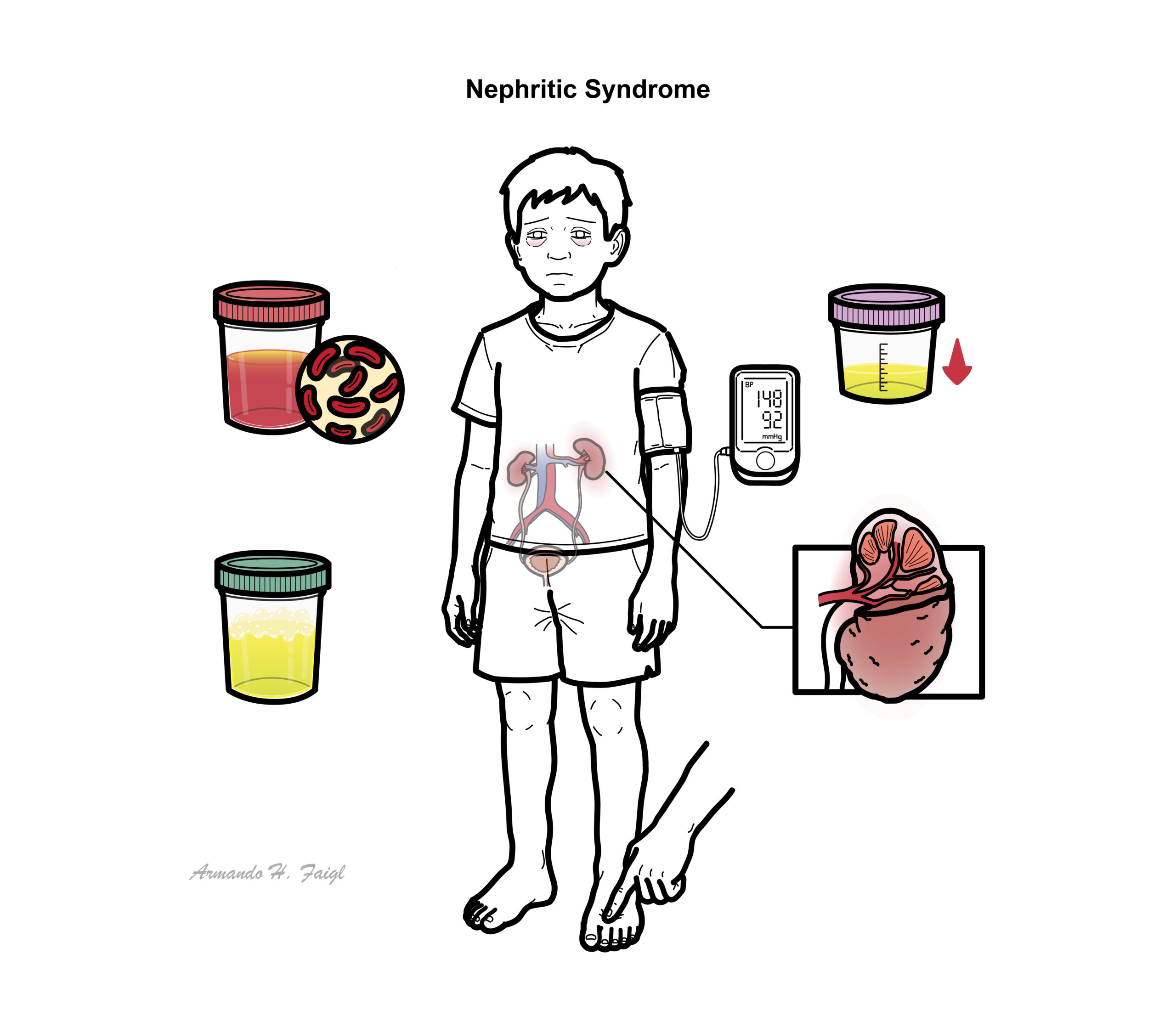
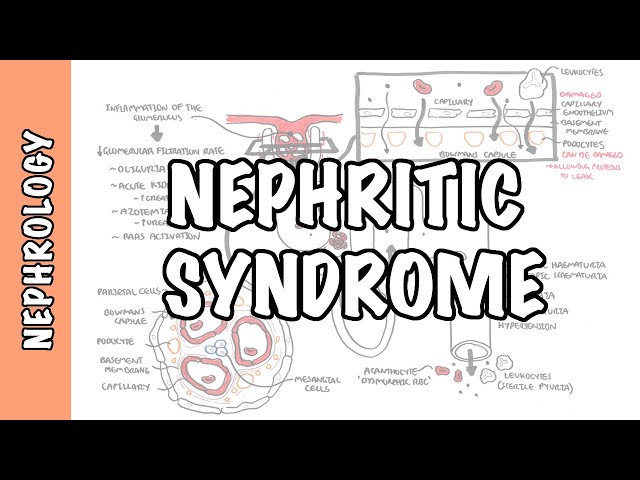
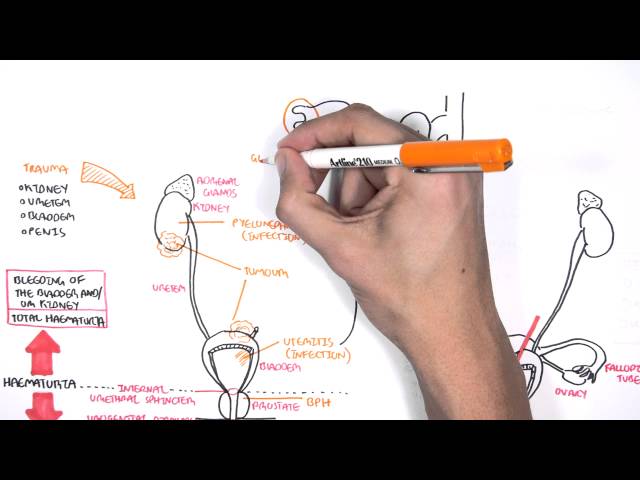
Nephritic syndrome is one of two clinical patterns seen in glomerular disease. Nephritic Syndrome is characterised by haematuria, proteinuria, hypertension and oligouria. Nephritic syndrome can be further classified based on light microscopic features:
- Focal
- Diffuse
Definition
Glomerular Disease: Disease effecting the glomerulus. Glomerular disease reduces the ability of the kidneys to maintain a balance of certain substances in bloodstream
Nephritic Syndrome: Group of symptoms that occur with some disorders that cause swelling and inflammation of the glomeruli in the kidney, or glomerulonephritis. Acute nephritic syndrome is often caused by an immune response triggered by an infection or other disease.
Nephrotic Syndrome: kidney disease, especially when characterized by oedema and the loss of protein from the plasma into the urine due to increased glomerular permeability
Acute Kidney Injury: Characterized by abrupt deterioration in kidney function, manifested by an increase in serum creatinine level with or without reduced urine output. The spectrum of injury ranges from mild to advanced, sometimes requiring renal replacement therapy. The diagnostic evaluation can be used to classify acute kidney injury as prerenal, intrinsic renal, or postrenal.
Chronic Kidney Failure: Defined as the presence of kidney damage, manifested by abnormal albumin excretion or decreased kidney function, quantified by measured or estimated glomerular filtration rate (GFR), that persists for more than three months.
Classification
The page will focus on four common nephritic syndromes towards the end:
- IgA nephropathy
- Lupus Nephritis
- Post-infectious Glomerulonephritis
- Anti-GBM disease
Anatomy and Physiology
The kidneys are retroperitoneal organs (at the back of abdomen) and lies between T11 and L3, the left kidney lying slightly higher than the right kidney.
The kidney is surrounding by an outer membrane called the renal capsule. The outer layer of the kidney is the renal cortex where most of the nephrons are situated. The inner layer is the renal medulla which contain renal pyramids. The renal pyramids drain urine into the minor – major calyces before draining to the renal pelvis then the ureter.
Arterial blood supply
- Abdominal Aorta → Renal arteries
- The renal artery branches into smaller and smaller arteries until finally becoming afferent arterioles which supply individual glomeruli
The Nephrons are functional units of the kidney. There are millions of nephrons. A nephron is made up of:
- Bowman’s capsule – afferent arteriole enters the bowmans capsule and forms t and efferent exits
- Proximal convoluted tubule
- Loop of Henle (descending and ascending)
- Distal convoluted tubule
- Collecting ducts
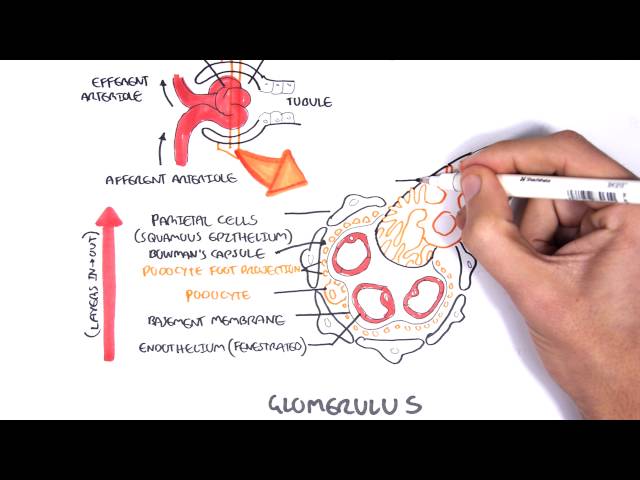
The glomerulus is a network of blood vessels that are located within the bowman’s capsule. To understand the cause, pathophysiology and effect of glomerulonephritis it is important to learn the layers of the glomerulus + bowman’s capsule.
From inside out the layers are (as depicted in the image above):
- Endothelial cells of the blood vessels
- Glomerular basement membrane
- Podocytes
- Bowman’s space
- Parietal cells.
- In the center of the glomerulus are the mesangial cells which help in regulating blood flow the glomeruli.
Clinical Manifestations
- Haematuria
- Proteinuria
- Oligouria – with signs of salt and water retention
- Hypertension
Pyuria can also be conspicuous in nephritic patients, particularly in inflammatory forms of glomerulonephritis such as poststreptococcal glomerulonephritis. However, pyuria is never the sole manifestation of nephritic urine sediment.
- IgA nephropathy and Henoch–Schönlein purpura
- Lupus nephritis
- Post-infectious GN
- Anti-GBM disease
- ANCA-associated vasculitis
- Mesangiocapillary GN (MCGN)
Investigations
- Dipstick urine for haematuria, proteinuria.
- Urine microscopy for red blood cell morphology, casts
- Amount of proteinuria variable
- eGFR
- EUC
- FBC
- Bone profile
- LFT
- Acute phase markers (CRP, ESR).
- Immunological and serological (‘nephritic’) screen
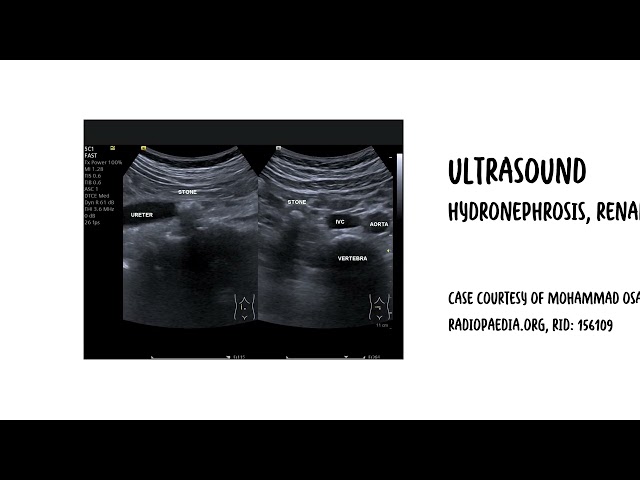
- Ultrasound of kidneys.
- Renal biopsy
Treatment
- Salt and water restriction – It is vital to correctly assess volume status
- Control Blood Pressure
- Diuretics
- ACE inhibitors or ARBs
- Immunosuppressive therapy – tailored to specific type of nephritic syndrome (based on histology)
- Corticosteroids
- Cyclophosphamide
- Chlorambucil
- Calcineurin inhibitors
- Azothiopurine
- Ritiximab
IgA Nephropathy
Overview
The most common primary glomerulonephritis in the world, affecting an estimated 1.5% of the population. Often presents with haematuria in the 2nd and 3rd decades. Occurs due to IgA immune complex deposition in glomerular mesangium and capillary wall, associated complement 3 activation and IgG binding and resulting glomerular destruction. Damage results in chronic microscopic or recurrent macroscopic haematuria, often after acute URTI or gastroenteritis
Previously considered relatively benign, it is now recognized that 30– 50% will progress to significant CKD over time.
- Liver cirrhosis and other liver disease (due to reduced IgA removal by Kuppfer cells)
- Coeliac disease (excessive IgA production against gliadin protein in gluten, form gliadin-antigliadin immune complexes, up to 30% coeliacs develop IgA nephropathy.
- HIV (abnormal IgA produced, form immune complexes)
- Family history
Clinical Presentation
- Painless Haematuria
- <2days after URTI of gastrointestinal infection
- Usually brown colour
- Oligouria or dysuria
- Abdominal or flank pain
- Fever
- Malaise
- Myalgia
- Hypertension
- Purpuric skin rash – IgA nephropathy associated with Henoch–Schönlein purpura
Recurrent gross haematuria is the hallmark of IgA nephropathy. Closely related to IgAN is Henoch–Schönlein purpura (HSP), a condition that occur in children. HSP is a small vessel systemic vasculitis characterised by IgA deposition in affected blood vessel, with kidney biopsy findings usually indistinguishable from IgAN.
Pathology and Diagnosis
- Light microscopy: Mesangial cell proliferation with matrix expansion
- Immunoflourescence: IgA, IgG and complement 3 protein deposition on mesangial cells
- Electron microscopy: Mesangial/paramesangial electron-dense deposits of immune complexes
Treatment for IgA nephropathy is only required with persistent proteinuria, recurrent haematuria, mild to moderate findings on renal biopsy and evidence of decreased GFR. Isolated haematuria does not require treatment
- ACE inhibitors
- Statin
- Omega 3 fish oil
- Immunosuppresive therapy (usually for high-risk patients) – Corticosteroirds, cyclophosphamide
- Dietary modification
- Renal replacement therapy – for end stage kidney disease
Pharmacology
Cyclophosphamide
- Chronic Kidney Disease
- End stage renal disease
- Acute renal failure
- IgA nephropathy is benign in majority of patients but up to 30% of patients progress to end stage renal disease within 10 years.
- Patients with minimal proteinuria or minimal renal dysfunction might undergo long-term remission periods
Post-Infectious glomerulonephritis
Overview
Post-infectious glomerulonephritis is classically associated with streptococcal infection, but infection of almost any cause may be associated with an acute nephritic syndrome and a diffuse proliferative GN on kidney. Post-infectious glomerulonephritis is an immune complex-mediated GN that usually occurs in childhood (age <7 years) following (usually) a upper respiratory tract infection (Tonsilitis, pharyngitis) but also impetigo, otitis media and cellulitis 1-2 weeks earlier.
Clinical presentation
- Frank haematuria
- Oliguria
- Oedema
- ↑BP
- Pulmonary oedema
- Acute Kidney Injury
- Loin Pain (may be bilateral)
The most common Post-infectious glomerulonephritis is Post-streptococcal glomerulonephritis. Other causes include: streptococcal pneumococcal, meningococcal, salmonella, mycobacterial, syphilis. Viruses: influenza B, mumps, rubella, coxsackie, hepatitis B, EBV, CMV. Fungi: candida, Coccidioides, Histoplasma. Parasites: malaria, filiariasis, toxoplasmosis, or schistosomiasis.
- As summarised above in the investigation section of nephritic syndrome
- Anti-streptolysin O titre
- Anti-DNAase B (for group A streptococci)
Pathology/Diagnosis
- Light Microscopy – Diffuse proliferative changes with hypercellularity. Extensive neutrophilic infiltration and red cell casts. Crescentic change and frank necroses are unusual
- Immunoflorecence – IgG and C3 deposition in diffuse granular pattern in both the mesangium and glomerular capillary walls
- Electron Miscroscopy – ‘dome-shaped’, electron-dense deposits in the subepithelial aspects of the capillary walls, with endothelial cell swelling
Course of Post-Infectious Glomerulonephritis
- Generally is self-limiting. It is associated with a full renal recovery (even after AKI).
- Resolution usually begins after 7–10 days.
- Recurrence is rare.
- Urinary abnormalities may persist for many years after recovery
- Proteinuria, ↑ BP, and CKD also occur, so patients should be offered follow-up
Treatment
- Ensure underlying infection has been resolved
- Restrict salt and fluid (~500-1000mL/day)
- Antihypertensive (Frusomide or ACE inhibitors)
- Renal replacment
Lupus Nephritis
Overview
Renal involvement is common in systemic lupus erythematosus (SLE). There are several types of renal disease in SLE (most commonly, immune complex-mediated glomerular disease), which are usually differentiated with a renal biopsy.
Pathophysiology The pattern of glomerular injury seen in systemic lupus erythematosus (SLE) (and in other immune complex-mediated glomerular diseases) is primarily related to the site of formation of the immune deposits, which are primarily due to anti-double-stranded DNA antibodies (anti-dsDNA, or anti-DNA).
Classification
- Class I disease (minimal mesangial lupus nephritis) is characterized by mesangial immune deposits that are identified either by immunofluorescence alone or by immunofluorescence and electron microscopy
- Class II disease (mesangial proliferative lupus nephritis) is characterized by mesangial hypercellularity or mesangial matrix expansion on light microscopy.
- Class III disease (focal lupus nephritis) is defined by light microscopic appearance of endocapillary or extracapillary glomerulonephritis that involves fewer than 50 percent of glomeruli.
- Class IV (diffuse lupus nephritis) is defined by more than 50 percent of glomeruli displaying endocapillary with or without extracapillary glomerulonephritis.
- Class V (lupus membranous nephropathy) is characterized by diffuse thickening of the glomerular capillary wall on light microscopy and by subepithelial immune deposits on immunofluorescence or electron microscopy.
- Class VI disease (advanced sclerosing lupus nephritis) is characterized by global sclerosis involving more than 90 percent of glomeruli.
Anti-GBM
Overview
Anti-GBM antibody disease is a disorder in which circulating antibodies are directed against an antigen intrinsic to the glomerular basement membrane (GBM), thereby resulting in acute or rapidly progressive glomerulonephritis that is typically associated with crescent formation.
Patients with anti-GBM antibody disease usually present with rapidly progressive glomerulonephritis: acute renal failure, nephritic urine sediment, and non-nephrotic proteinuria. Pulmonary involvement (alveolar haemorrhage) is present in 40 to 60 percent of patients
Diagnosis
- Presence of anti-GBM antibodies
- Kidney biopsy, unless contraindicated, since demonstration of linear deposits of IgG (which represent binding of anti-GBM antibodies to the GBM)
Other investigations
- Antineutrophil cytoplasm antibodies



Members only discussions coming soon…