

Interstitial Lung Disease
Overview
Interstitial Lung Disease is also known as Diffuse Parenchymal Lung Disease (DPLD). A Heterogeneous collection of restrictive lung conditions (>100) that primarily (or at least initially) disrupt the pulmonary interstitium. Interstitial Lung Disease (ILD) are grouped together as they share similar pathophysiological mechanisms, clinical features and radiological findings.
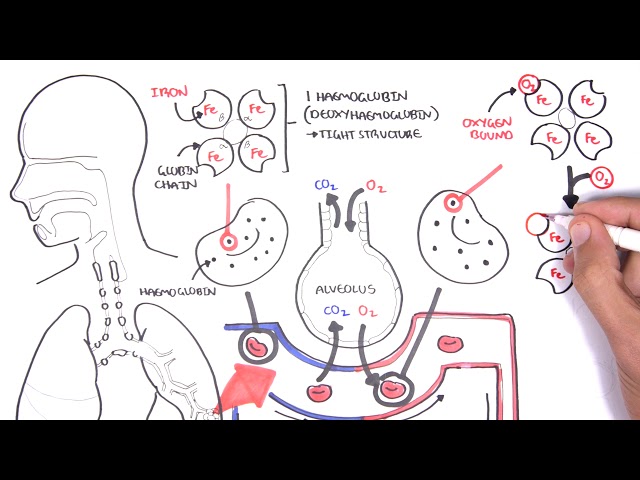
Pulmonary Interstitium: Intervening structural tissue between the alveoli space and pulmonary capillaries. Consists of the basement membrane of endothelial and epithelial cells, collagen fibres, elastic tissue, proteoglycans, fibroblasts, mast cells and mononuclear cells.
Definition
Interstitial Lung Disease: Includes a diverse group of respiratory conditions characterised by inflammation and fibrosis of the interstitium. Worsening hypoxia and respiratory failure may develop with disease progression. The disease is considered a restrictive lung disease
Restrictive Lung Disease: Unable to fully fill the lungs with air. Their lungs are restricted from fully expanding.
Chronic Obstructive Lung Disease: A serious, progressive and disabling condition that limits airflow in the lungs. It includes emphysema and chronic bronchitis. Strongly associated with smoking
Pneumonia: Infection and Inflammation of the Lung paranchyma
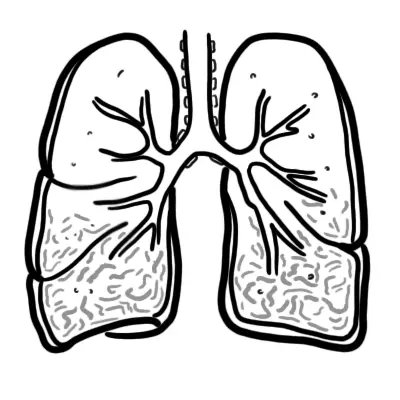
Respiratory Anatomy & Physiology
Simplified Anatomy of the alveolus and pulmonary capillary. Below is a diagram of the Interstitial Lung Parenchyma from a cross section. Many cells are found here:
- Type I Pneumocytes
- Type II Pneumocytes which produce surfactant
- Alveolar macrophages for pulmonary defences
- Interstitial fibroblasts, that make up the ECM and produce collagen and other matrix
Aetiology & Risk Factors
Age: There is significant variation in the age of onset between different ILDs.
- Young adult, Middle-aged – Sarcoidosis, Pulmonary Lagnerhan cells histocytosis
- Elderly – Idiopathic pulmonary fibrosis
Gender: There is significant variation in the gender distribution of different ILDs.
- Women
- Lymphangioleiomyomatosis
- Lymphocytic interstitial pneumonitis
- Hermansky-Pudlak syndrome
- Men
- Rheumatoid Arthritis
- Pneumoconiosis
Pathology
General changes seen in interstitial lung disease. There is proliferation of interstitial fibroblasts and recruitment of fibrocytes into the interstitium (Extracellular matrix [ECM]). These cells begin secreting ECM such as collagen and other fibers. The cells also convert and activate becoming myofibroblasts which further produces ECM. Prolonged fibroblast activity leads to fibrosis a hallmark of interstitial lung disease and restrictive lung pattern.
Clinical Manifestation
Clinical Presentation
- Dyspnoea
- Spontaneous pneumothorax – a characteristic finding of certain ILDs (e.g. pulmonary Langerhans cell histiocytosis)
- Dry Cough
- Haemoptysis – Diffuse alveolar haemorrhage
- Wheeze (uncommon)
- Chest Pain (uncommon but perhaps in sarcoidosis)
- Extrapulmonary Manifestations: May be suggestive of connective tissue disease.
| Onset of Symptoms: The duration of symptoms prior to presentation differs between ILDs and therefore may assist in narrowing down the diagnosis. | |
| Acute (days to weeks) | Acute eosinophilic pneumonia, Hypersensitivity pneumonitis |
| Subacute (weeks to months) | Sarcoidosis |
| Chronic (Months to years) | Idiopathic pulmonary fibrosis, pulmonary Langerhans cell histiocytosis |
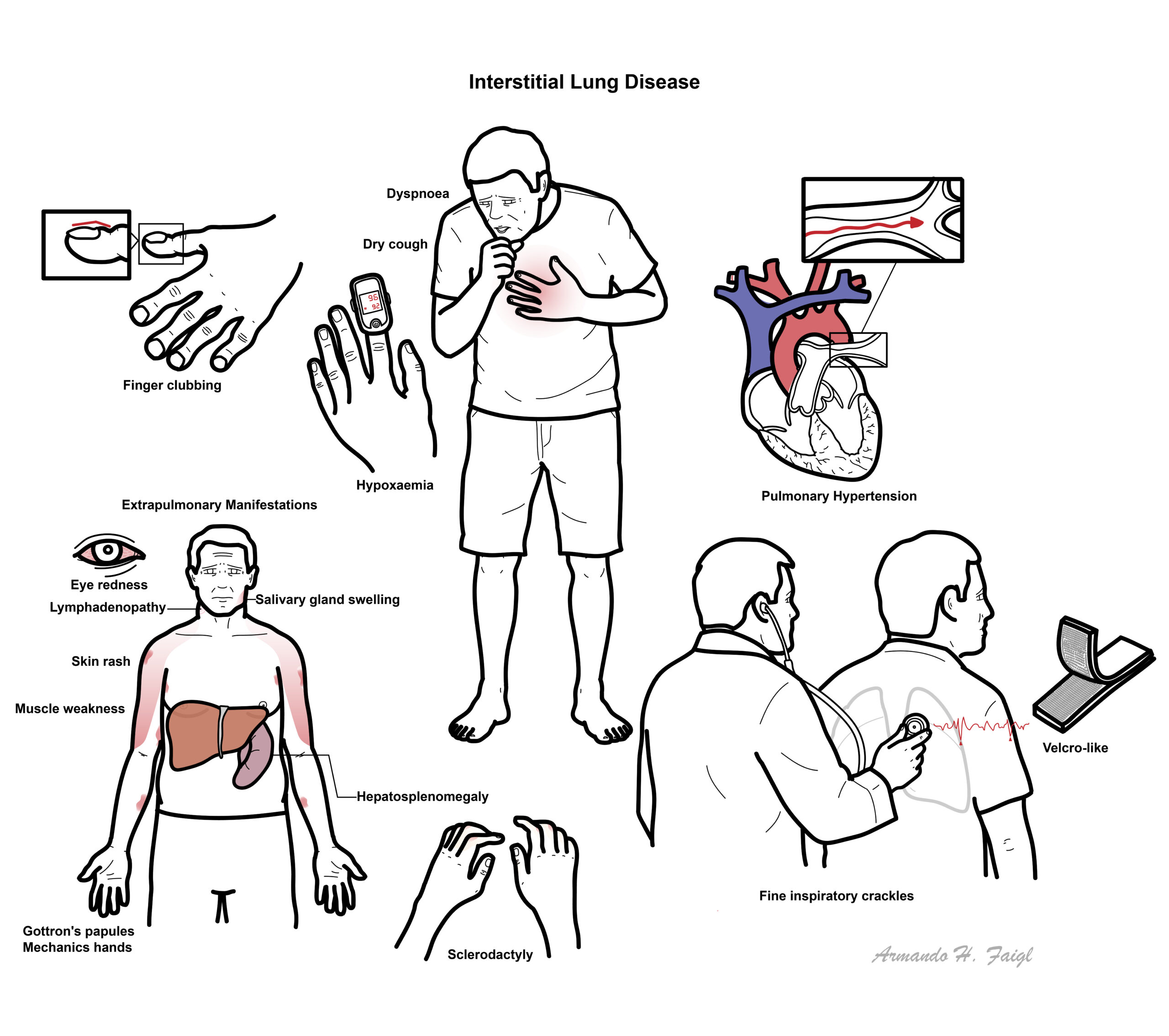
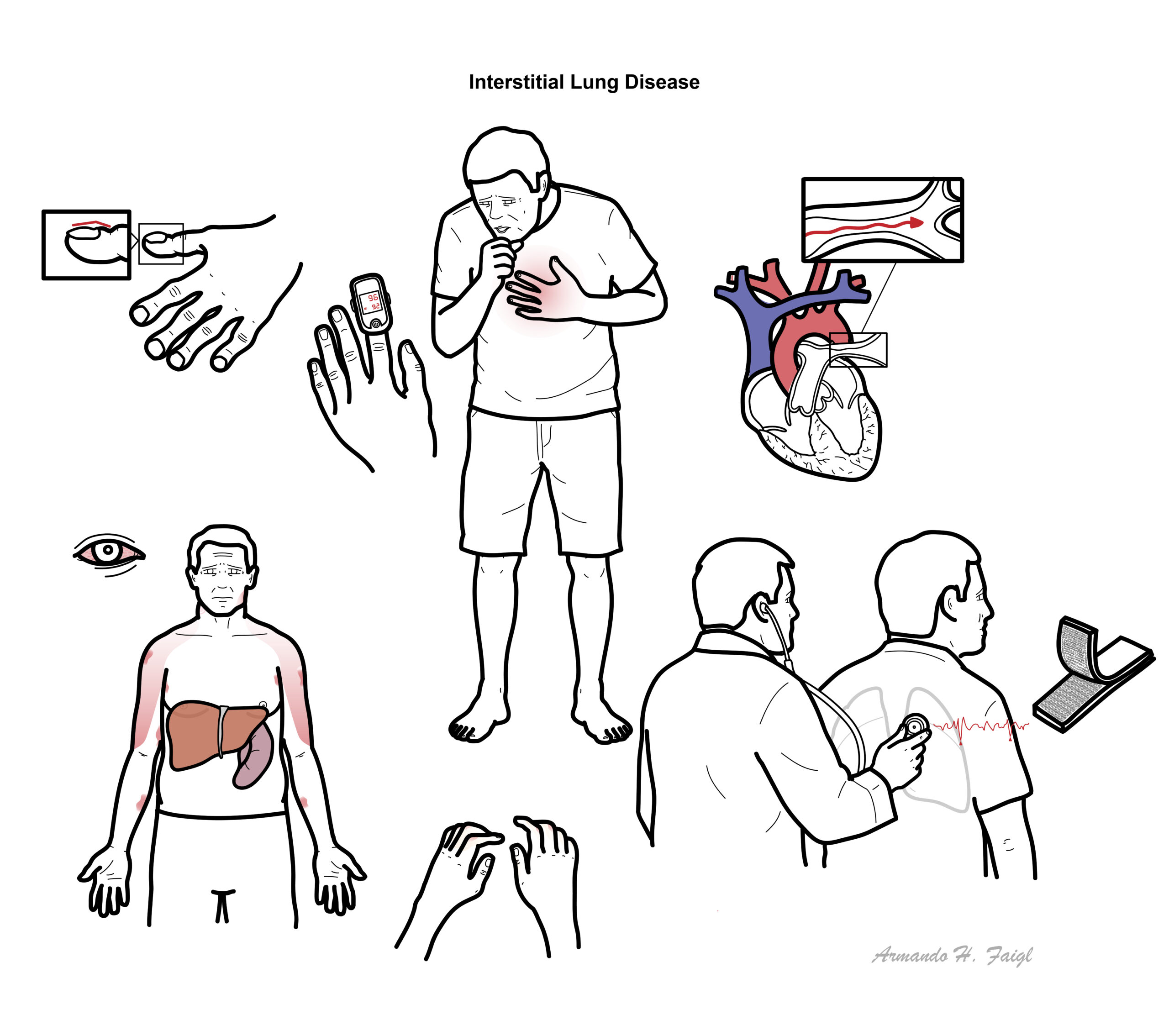
Examination
- Dyspnoea
- Clubbing
- Crakles (rales)
- Wheeze
- Pulmonary hypertension (severe)
- Systemic findings
- Skin Changes
- Eye Changes
- Salivary Gland enlargement
- Peripheral Lymphadenopathy
- Hepatosplenomeagly
- Pericarditis
- Myositis
Diagnosis
- Viral Pneumonia
- Tuberculosis
- Fungal infection
- Leukaemia
- Lymphoma
- Pulmonary Oedema
- Aspiration Pneumonitis
- FBC
- ABG
- LFT
- EUC
- ANA – if SLE suspected
- C-ANCA – if suspect hypersensitivity pneumonitis
- Chest X-ray – usually normal
- Small lung volume
- Reticularnodular shadowing
- Hilar / mediastinal lymphanenopathy
- CT
- Reticularnodular shadowing
- Ground glass changes
- Honeycomb cysts
CT changes found in ILD include reticularnodular shadowing, ground glass changes and honeycomb cysts.
- Spirometry – Restrictive Pattern
- ECG – Pulmonary Hypertension?
- Lung Biopsy – Transbronchial or Surgical Lung Biopsy
ECG Should be performed in all suspected cases of ILD to look for evidence of pulmonary hypertension.
Classification
Due to the extensive number of subtypes, correct diagnosis of ILDs/DPLDs is challenging but essential to ensure optimal treatment and prognosis. Classification is informed by the patients history of exposure, clinical features, serology, radiological pattern and lung biopsy
The major subgroups of ILD are broadly defined as:
- Diffuse Parenchymal Lung Disease of known cause
- Drug induced
- Connective tissue disease associated (i.e Rheumatoid arthritis)
- Idiopathic interstitial pneumonia
- Idiopathic Pulmonary Fibrosis
- Acute interstitial pneumonia
- Lymphocytic interstitial pneumonia
- Granulomatous Diffuse Parenchymal Lung Disease
- Sarcoidosis
- Hypersensitivity pneumonitis
- Other Diffuse Parenchymal Lung Disease
- Pulmonary Lagnerhan cells histocytosis
Sarcoidosis is a multi-system disease that results in pulmonary fibrosis in up to 20% of patients. Aetiology is unknown. However, well-formed granulomas, the characteristic pathological lesion, are thought to form in response to a variety of environmental stimuli (eg infection, inorganic particles and certain chemicals).
Connective tissue disease associated ILD This includes scleroderma, systemic lupus erythematosus, rheumatoid arthritis, dermatopolymyositis and mixed connective tissue disease. Immune-mediated inflammation of the lung interstitium can occur, sometimes progressing to irreversible fibrosis if it is left untreated.
Complications
| Common Comorbidities in Interstitial Lung Disease |
| GORD/GERD |
| Osteoporosis |
| Infections |
| Obstructive sleep apnea |
| Pulmonary hypertension |
Restrictive vs. Obstructive
The hallmark of restrictive lung disease is decreased lung capacities, particularly the TLC but also the vital capacity.
In both obstructive and restrictive lung disease, the FEV1 is decreased, the FEV1 /FVC is decreased in obstructive processes and normal in restrictive processes.






Members only discussions coming soon…