

Marfans Syndrome
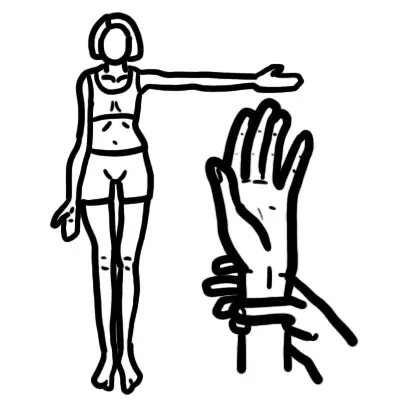
Overview
Marfan’s Syndrome is one of the most common inherited disorders of connective tissue. Marfan syndrome is a hereditary disease affecting connective tissues in the body, resulting in symptoms such as aortic dissection and musculoskeletal deformities. However, no family history of Marfan’s does not exclude the diagnosis as 30% can occur through gene mutation.
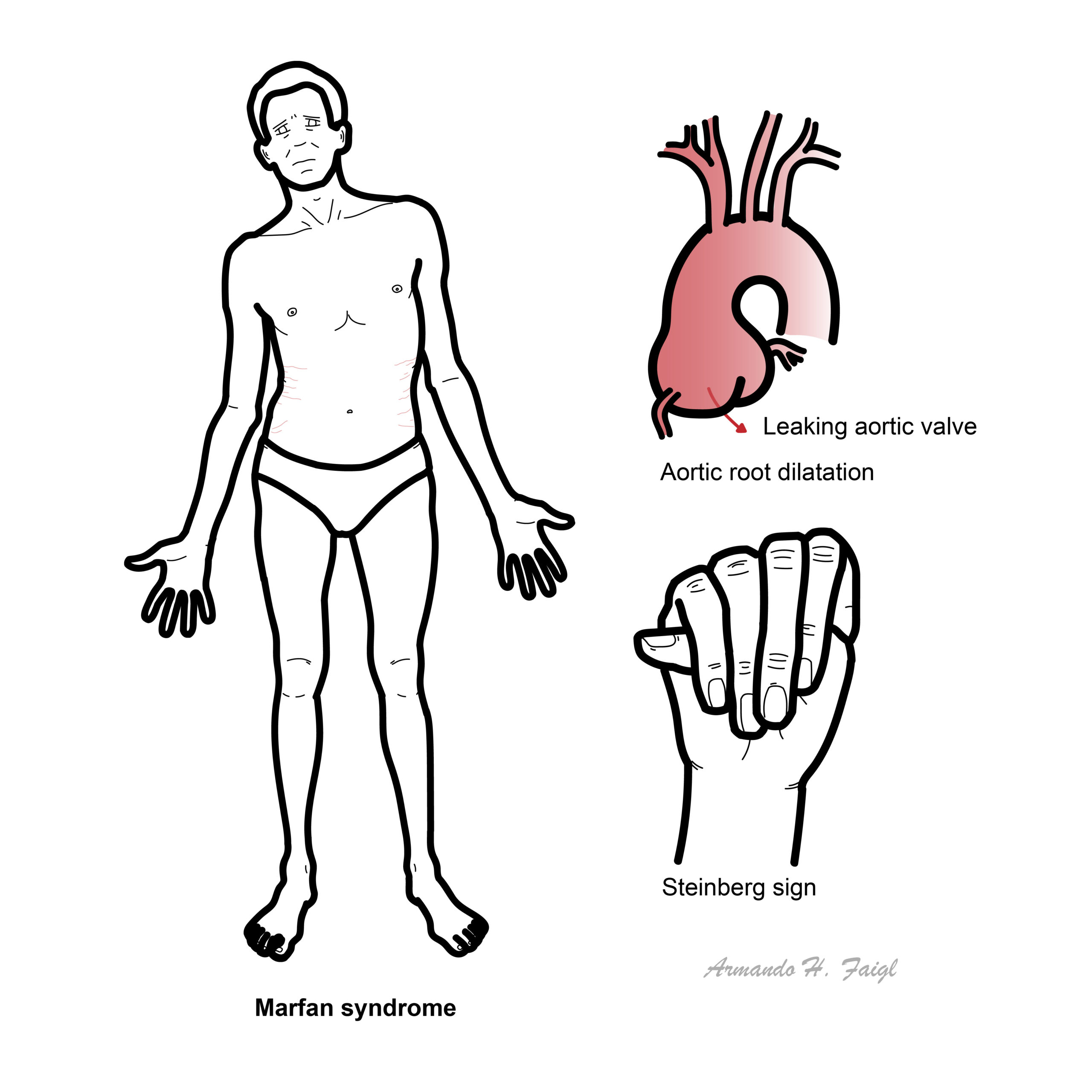
Definition
Marfan syndrome: Autosomal dominant disease as a result of mutation in the FBN1 gene. FBN1 gene mutation results in decreased production of fibrillin microfibrils and increased production of TGF- beta, which produces various clinical symptoms and signs.
Fibrillin microfibrils are large glycoproteins that form part of the extracellular matrix. Fibrillin microfibrils endow connective tissues with long-range elasticity.
Antoine Bernard- Jean Marfan (1958-1942) a french paediatrician described a hereditary disorder of connective tissue in a 5 yo girl with disproportionaly long limbs.
Aetiology & Risk Factors
Aetiology
Fibrillin 1 (FBN1) gene mutation, located on chromosome 15
- 75% – Inherited the mutated FBN1 gene from one or both parents, autosomal dominant
- 25% Sporadic, FBN1 gene underwent mutation in parent germ cells
- Family history of Marfan syndrome
- Family history of aortic dissection or aneurysm
- High parental age (Weak)
Pathophysiology
The fibrillin 1 gene is crucial in the production of fibrillin, a glycoprotein which stabilises crosslinks of microfibril tissues found in connective tissues, such as arteries and skin. Fibrillin either bind microfibril strands to form non-stretchable tissues (such as tendons), or binds microfibril strands with elastin to form elastic fibres (such as arteries). It also sequesters transforming growth factor beta (TGF-B), and promotes connective tissue growth.
With the deformed or deficiency in fibrillin proteins due to the gene mutation, connective tissues loss mechanical integrity.
Cardiovascular
In the aorta, the lost of integrity in elastic connective tissue causes necrosis of the tunica media, resulting in aortic dilation, which in turns causes aortic valve insufficiency and heart
failure. There is also increased risk of aneurysm formation and dissection of the aorta, causing mortality related to Marfan syndrome.
Musculoskeletal
Marfanoid body habitus, scoliosis, kyphoscoliosis and arthropathy are common symptoms present in Marfan syndrome. Since less fibrillin is present for sequestering the TGF-B, there is a overabundance of such substances in the systemic circulation, therefore other tissues such as epiphyseal plate responds to the constant stimulation of TGF-B, resulting in over-production of long bones.
Opthalmopathy
Increased risk of lens dislocation and retinal detachment due to insufficient connective tissue integrity.
Dermatological
Striae may be seen due to loosening of connective tissues in the dermal layer.
Lungs
Possible bulla formation in the lungs due to lack of mechanical integrity in lung elastic tissues increases risk of spontaneous pneumothorax.
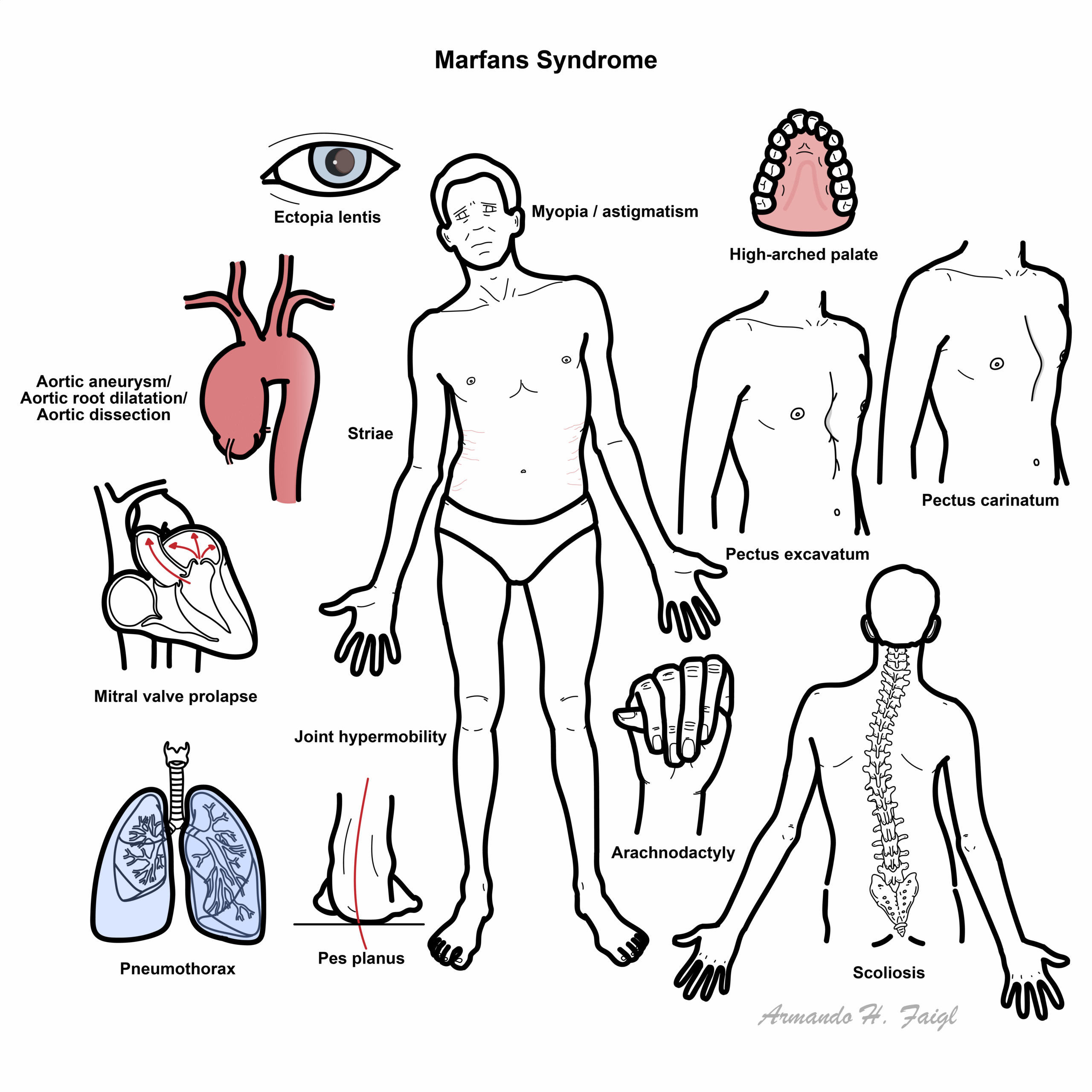
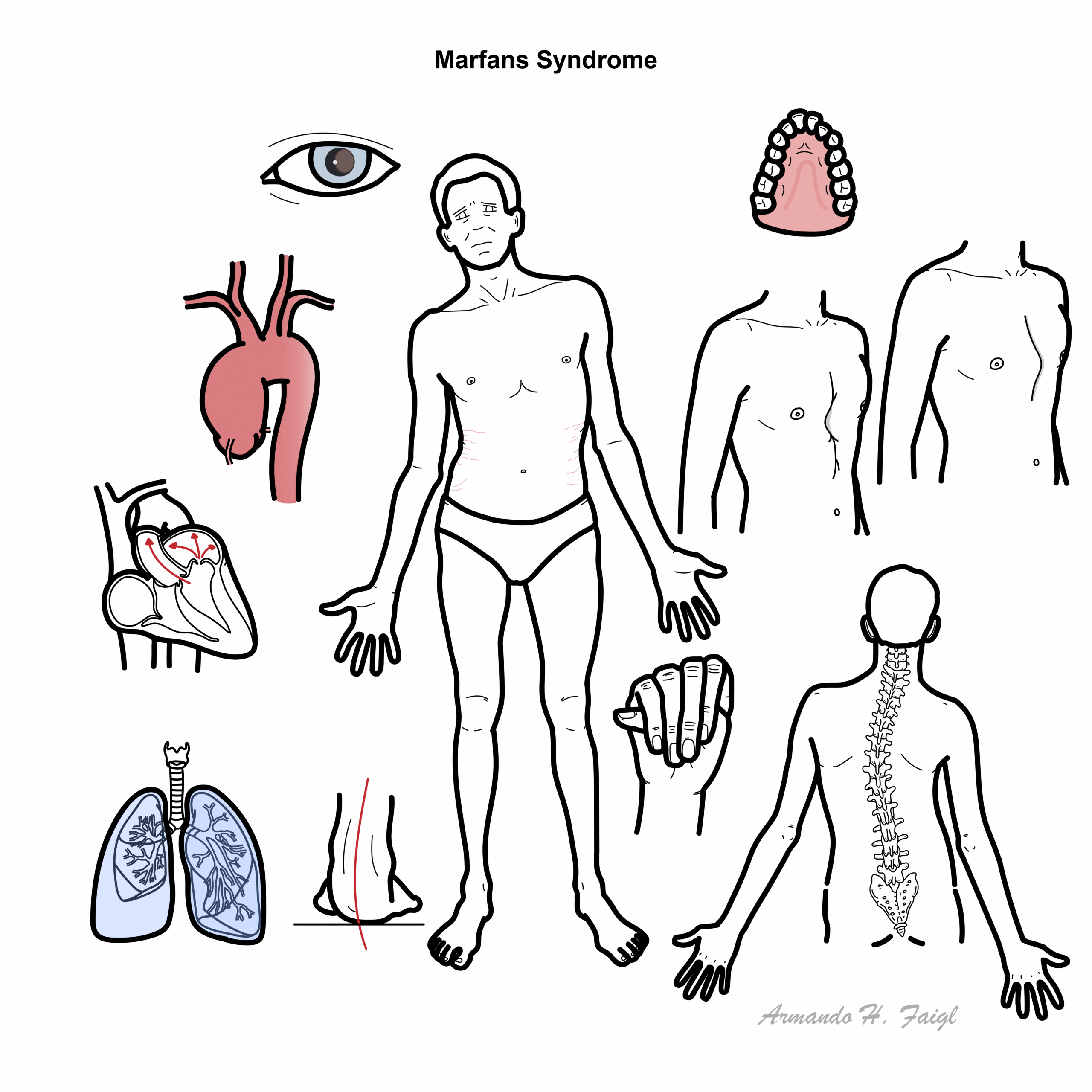
Clinical Manifestation
Signs and symptoms of Marfan syndrome varies among patients, they are not generally not presented at birth and develops overtime with age. For presentations at birth, it is regarded to as neonatal Marfan syndrome.
- Body habitus / musculoskeletal
- High stature, wide arm span – Marfanoid body habitus
- Chest deformity
- Pectus excavatum (Funnel chest)
- Pectus carinatum (Pigeons chest)
- Arachnodactyly (Long and thin digits)
- Scoliosis
- Joint hypermobility
- High arched palate
- Flat feet (pes planus)
- Cardiopulmonary
- Aorta dilatation – causing aortic valve insufficiency
- Aortic aneurysm
- Aortic dissection
- Mitral valve regurgitation
- Cystic medial necrosis (tunica media necrosis of the aorta)
- Pneumothorax due to lung bullae upper lobes
- Ophthalmology
- Ectopia lentis
- Dislocated/subluxed lens
- Myopia with astigmatism (blurred vision as a result of irregularly shaped cornea or lens)
Two cardinal features of Marfan Syndrome is aortic root dilatation and ectopia lentis. This is used in Diagnosis together with family history and confirmed FBN1 mutation.
Diagnosis
<20 years old
- Potential Marfans Syndrome
>20 years old
- MASS – Myopia, Mitral valve prolapse, Aortic root dilatation, Aortic aneurysm syndrome, Striae, Skeletal
- Loeys Dietz syndrome
- Hypertelorism, cleft palate/bifid uvula, arterial totuosity, aneurysm and dissection
- Ectopia lentis syndrome
- Ehlers Danlos syndrome (vascular type)
- Marfanoid hypermobility syndrome
Ehlers Danlos Syndrome disorder in connective tissue (collagen), characterised by joint hypermobility, elastic skin, skin thinning and bleeding disorders.
Investigation and Diagnosis
- Echocardiogram
- Chest X-ray
- CT scan of thorax
- Fibrillin 1 mutation testing
- Slit lamp eye examination
- Ultrasound of abdomen
Ghent Diagnosis (simplified)
Absence of family history
- Aortic root dilatation + Ectopia lentis
- Aortic root dilatation + FBN1 gene mutation
- Aortic root dilatation + systemic score >7points
Presence of family history
- Family history + ectopia lentis
- Family history + systemic score >7points
- Family history + aortic root dilatation
Treatment
Treatment of Marfan syndrome is based on symptoms management and improvement of overall life quality of the patient. Each system affected by the FBN1 gene mutation can be
treated separatly.
- Cardiovascular
- Beta blocker
- Angiotensin II receptor blocker (ARB)
- Prophylaxis for endocarditis and anticoagulants for high risk procedures or surgeries
- Surgical treatment in cfor symptomatic aortic pathology
- Opthalmopathy – surgical for cataracts, myopia, retinal detachment
- Musculoskeletal
- Scoliosis/kyphoscoliosis : Orthopaedic bracing or surgery
- Pectus excavatum or pectus carinatum affecting cardiorespiratory system: Surgery
- Arthropathy: Physiotherapy, analgesics, surgery
Complications & Prognosis
- Cardiovascular
- Aortic dilation
- Acute aortic dissection/rupture
- Chronic aortic dissection
- Symptomatic aortic regurgitation
- Heart failure
- Infective endocarditis
- Respiratory
- Spontaneous pneumothorax
- Abdominal
- Symptomatic inguinal/incisional/abdominal hernia
- With appropriate medical and surgical intervention addressing the symptoms and complications of Marfan syndrome, the long term survival rate of most patients are good, most patients can live a normal lifespan with reasonable quality of life.
- Survival rate is significantly reduced by presentation of aortic dissection even with effective treatment.
References
- Child. A. and Tome. M. (2019). Marfan syndrome. BMJ Best Practice. https://bestpractice-bmj-com.wwwproxy1.library.unsw.edu.au/topics/en-gb/514. Last accessed 11 March 2019.
- Colledge, N. R., Walker, B. R., Ralston, S., & Davidson, S. (2010). Davidson’s principles and practice of medicine. Edinburgh: Churchill Livingstone/Elsevier.
- Groenink, M., Lohuis, T. A. J., Tijssen, J. G. P., Naeff, M. S. J., Hennekam, R. C. M., Van Der Wall, E. E., & Mulder, B. J. M. (1999). Survival and complication free survival in Marfan’s syndrome: implications of current guidelines. Heart, 82(4), 499-504.
- Kumar, V., Abbas, A. K., Aster, J. C., & Robbins, S. L. (2013). Robbins basic pathology. Philadelphia, PA: Elsevier/Saunders. Svensson, L. G., Crawford, E. S., Coselli, J. S., Safi, H. J., & Hess, K. R. (1989). Impact of cardiovascular operation on survival in the Marfan patient. Circulation, 80(3 Pt 1), I233-42.
- Wright. M. and Connolly. H. M. (2019). Genetics, clinical features, and diagnosis of Marfan syndrome and related disorders. Up To Date. https://www.uptodate.com/contents/genetics-clinical-features-and-diagnosis-of-marfan-syndr me-and-related-disorders?search=marfan%20syndrome&source=search_result&selectedTitl =1~150&usage_type=default&display_rank=1 Last accessed 11 March, 2019.
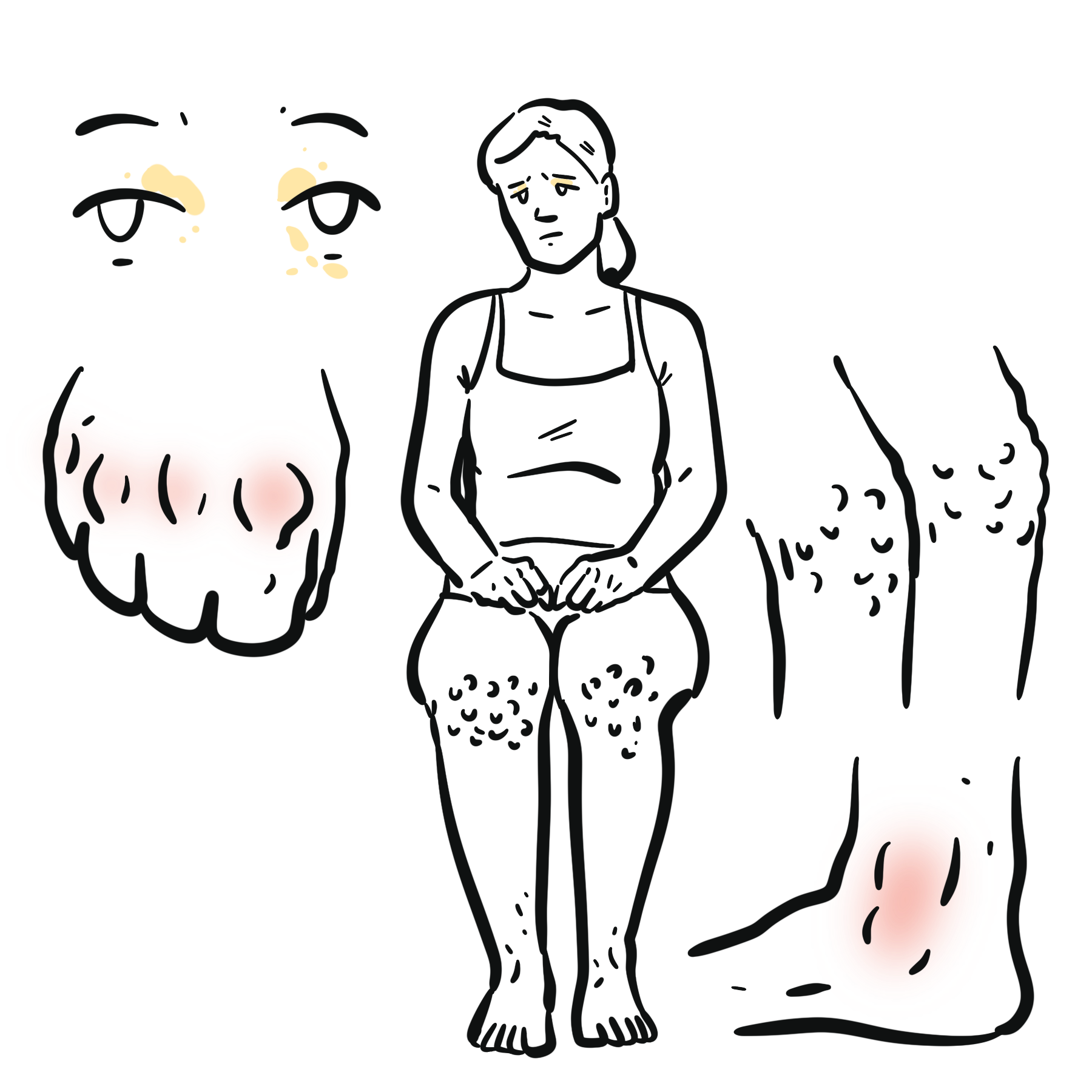










Members only discussions coming soon…