Overview
Overview Cystic fibrosis causes the dehydration of the mucus lumen causing thick sticky mucus to build up. This occurs mainly the lungs, causing productive cough and increasing rates in respiratory infections, and the pancreas causing pancreatic dysfunction. Cystic fibrosis occurs due to a mutation in the CFTR gene. Salty sweat is also another important characteristic of these patients.
| Definition Cystic Fibrosis (CF): The major cause of chronic debilitating pulmonary disease and pancreatic exocrine deficiency in the first three decades of life. It is characterized by the triad of chronic obstructive pulmonary disease, pancreatic exocrine deficiency, and abnormally high sweat electrolyte concentrations. Characteristic pancreatic changes give the disease its name. Clubbing: Increase in the angle between the nail and nail base of 180° or greater, and softening of the nail base to palpation. Although the condition can be familial, clubbing is uncommon in children, usually indicating chronic pulmonary, hepatic, cardiac, or gastrointestinal disease. Meconium ileus: Neonatal bowel obstruction of the distal ileum due to abnormally thick and impacted meconium. Unlike in the meconium plug syndrome, the meconium is abnormal in consistency. |
| Video (NOT COMPLETE): Cystic Fibrosis Overview |
Signs and Symptoms
The clinical presentation of cystic fibrosis defer in infants and children.
Infants
- Meconium ileus (bowel obstruction due to poo)
- Rectal prolapse
Children - Mainly failure to thrive and recurrent upper respiratory tract infection
- Chronic cough and wheezing
- FTT
- Pancreatic insufficiency (symptoms of malabsorption such as steatorrhea)
- Alkalosis and hypotonic dehydration
- Neonatal intestinal obstruction (meconium ileus)/Nasal polyps
- Clubbing of fingers/Chest radiograph with characteristic changes
- Rectal prolapse
- Electrolyte elevation in sweat, salty skin
- Absence or congenital atresia of vas deferens
- Sputum with S. aureus or P. aeruginosa (mucoid)
| Remember Cystic Fibrosis usually presents in childhood as recurrent lung infections that become persistent and chronic. |
Differential Diagnosis
- Failure to thrive
- Asthma
- Coeliac Disease
- GORD
- Immunodeficiency disorders
Investigation
- Infant have a Heel prick - Immunoreactive Trypsinogen (IRT) to detect cystic fibrosis
| Side note Heel prick is performed to screen for metabolic disease of the newborn inlcuding: galactosemia, cystic fibrosis, phenylketouria, sickle-cell disease and congenital hypothyroidism. |
- Sweat test - first line for diagnosis
- >60mmol/L
- Genetic screening - Type of CFTR mutation
Aetiology
Aetiology A mutation of the CFTR gene on the long arm of chromosome 7 is the cause of cystic fibrosis. The mutation of the CTFR gene results in a mutated CFTR protein which is a transported for Cl- ions. The severity of cystic fibrosis depends on the type of mutation found on the CFTR gene.
Pathophysiology
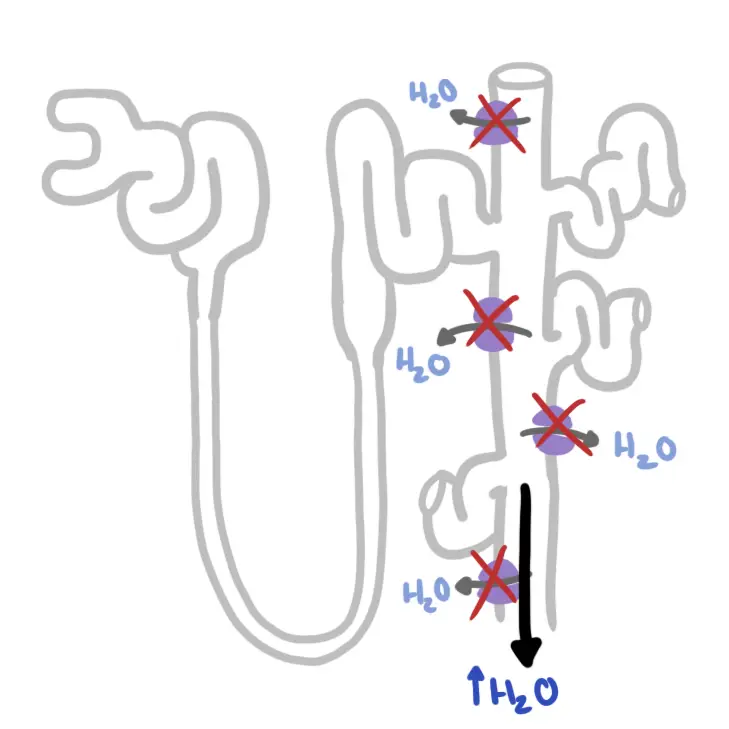
Pathophysiology Cystic Fibrosis is caused by mutated CTFR protein. The CFTR protein is found on the apical surface of many hollow tissues notably the lungs, pancreas, gastrointestinal tract and sweat glands.
The CTFR protein is a transporter for Cl-. A mutated CFTR protein can not transport Cl- properly and as a result this causes a disregulated ion distribution. Whereever Cl- exists Na+ will follow because of the electrochemical gradient. When Na+ move water tends to follow.
This basic principle is the cause of the signs and symptoms seen in Cystic Fibrosis
| Remember Cystic Fibrosis is characterized by the triad of chronic obstructive pulmonary disease, pancreatic exocrine deficiency, and abnormally high sweat electrolyte concentrations. |
Management
General
- High fat, high calorie diet
- Pancreatic enzyme supplement
- Vitamin supplementation (ADEK)
- Salt supplementation
- Pulmonary care
- physiotherapy
- shaking chest jackets
- Long-term antibiotics (inhaler)
- Bronchodilator
Management of upper respiratory tract infection
- Oral Antibiotics
- IV antibiotics
| Remember patients with CF are at risk of having recurrent URTI which can be caused by S. aureus, H. Influenzae, P. aerugenosa. |
Management of non-pulmonary complications
Complication and Prognosis
The complications of cystic fibrosis can be classified into respiratory, pancreatic, metabolic and medication side effects.
- Respiratory complications occur because of the thick mucous build up
- Infections
- Pancreatic dysfunction - the pancreas can not secrete its enzymes properly resulting in malabsorption and failure to thrive. Further, the pancreas can fail in both exocrine and endocrine function. When the endocrine function is involved diabetes mellitus may result.
Prognosis Cystic Fibrosis is a genetic disease for which there is no cure. However, the outlook for patients with this condition has greatly improved. In the past 50 years, the mean age of survival has risen from infancy/school age to almost 38 years old.