

Myelofibrosis
Overview
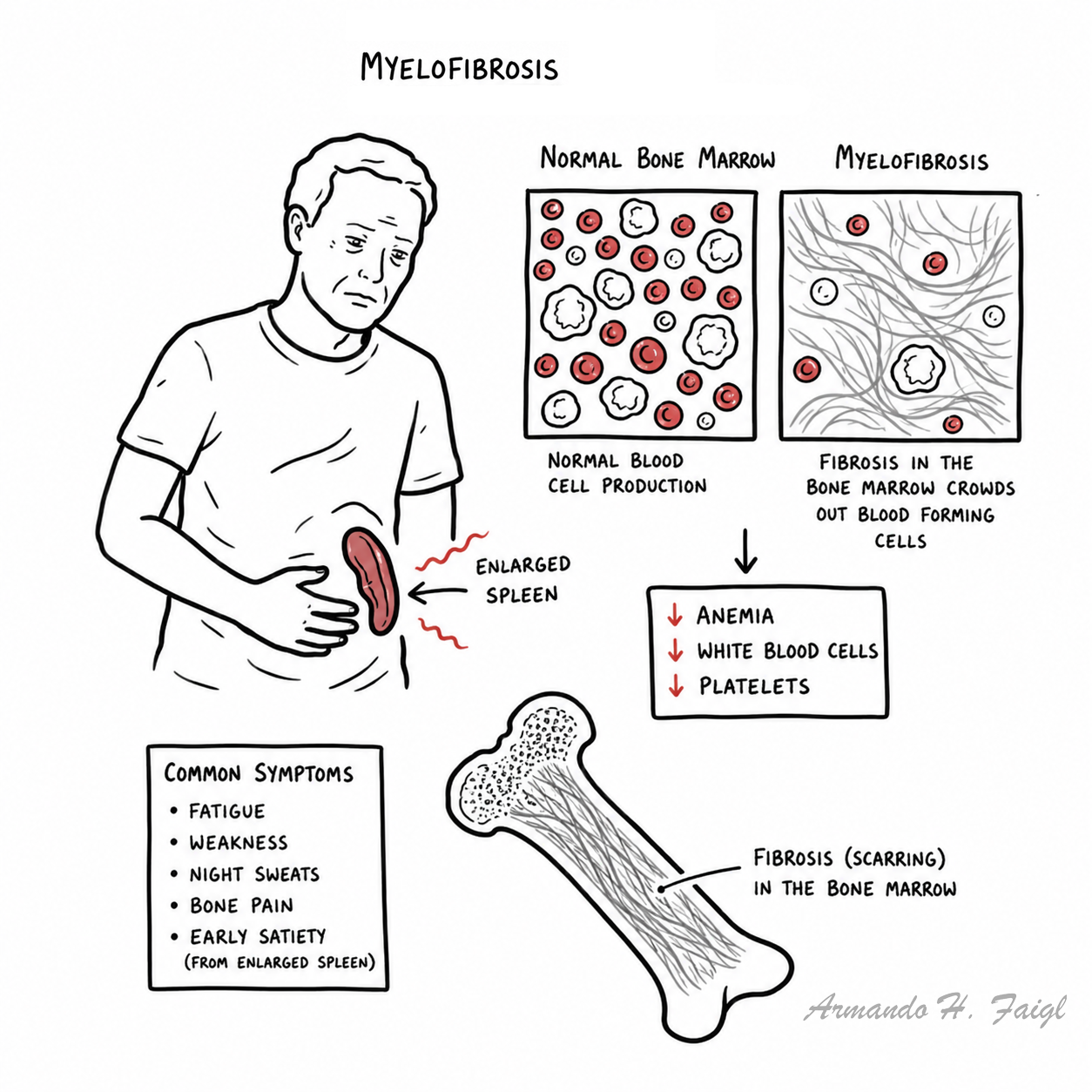
Myelofibrosis is a type of myeloproliferative diseasecharacterized by clonal myeloid expansion, followed by progressive fibrous connective tissue deposition in the bone marrow, resulting in bone marrow failure. Clonal evolution can also occur, with an increased risk of transformation to acute myeloid leukemia. In addition, disabling constitutional symptoms secondary to the high circulating levels of proinflammatory cytokines and hepatosplenomegaly frequently impair quality of life.
Definition
Myeloproliferative disorder: are clonal stem disorders characterised by leukocytosis, thromboytosis, erythrocytosis, splenomegaly, and bone marrow hypercelluarity. Includes myelofibrosis, essential thrombocytopaenia and polycythaemia
Primary Myelofibrosis: one of the chronic myeloproliferative disorders, where there is replacement of bone marrow with collagenous connective tissue and progressive fibrosis
Secondary Myelofibrosis: Myelofibrosis caused secondary to another disease such as essential thrombocytopnaeia or polycythaemia rubra vera
Essential Thrombocytopaenia: one of the chronic myeloproliferative disorders, in which too many platelets are produced in the bone marrow
Polycythaemia rubra vera: one of the chronic myeloproliferative disorders, which are collectively characterized by clonal proliferation of myeloid cells with variable morphologic maturity and hematopoietic efficiency.
Three main types of myeloproliferative disorders are PRV, ET and PMF.
Hypersplenism triad: splenomegaly, pancytopenia and normocellular bone marrow.
Aetiology & Risk Factors
Aetiology
Exact mechanism of disease is unknown but factors include:
- Chromosomal abnormality (ie. retinoblastoma gene mutation)
- JAK2 mutation (50%) → causes proliferation and survival of cells
- JAK/STAT mutation → causes proliferation and survival of cells
- MPL mutation → thrombopoietin receptor protein mutation → proliferation and survival of megakaryotes
- Thrombopoietin receptor gene mutation
- Overexpression of thrombopoietin
- Abnormal cytokine release (Growth factors) by cells in bone marrow, mainly megkaryotes → proliferation of fibroblasts → ↑collagen and fibrosis
- Radiation Exposure
- Age >65yo
Clinical Manifestation
General
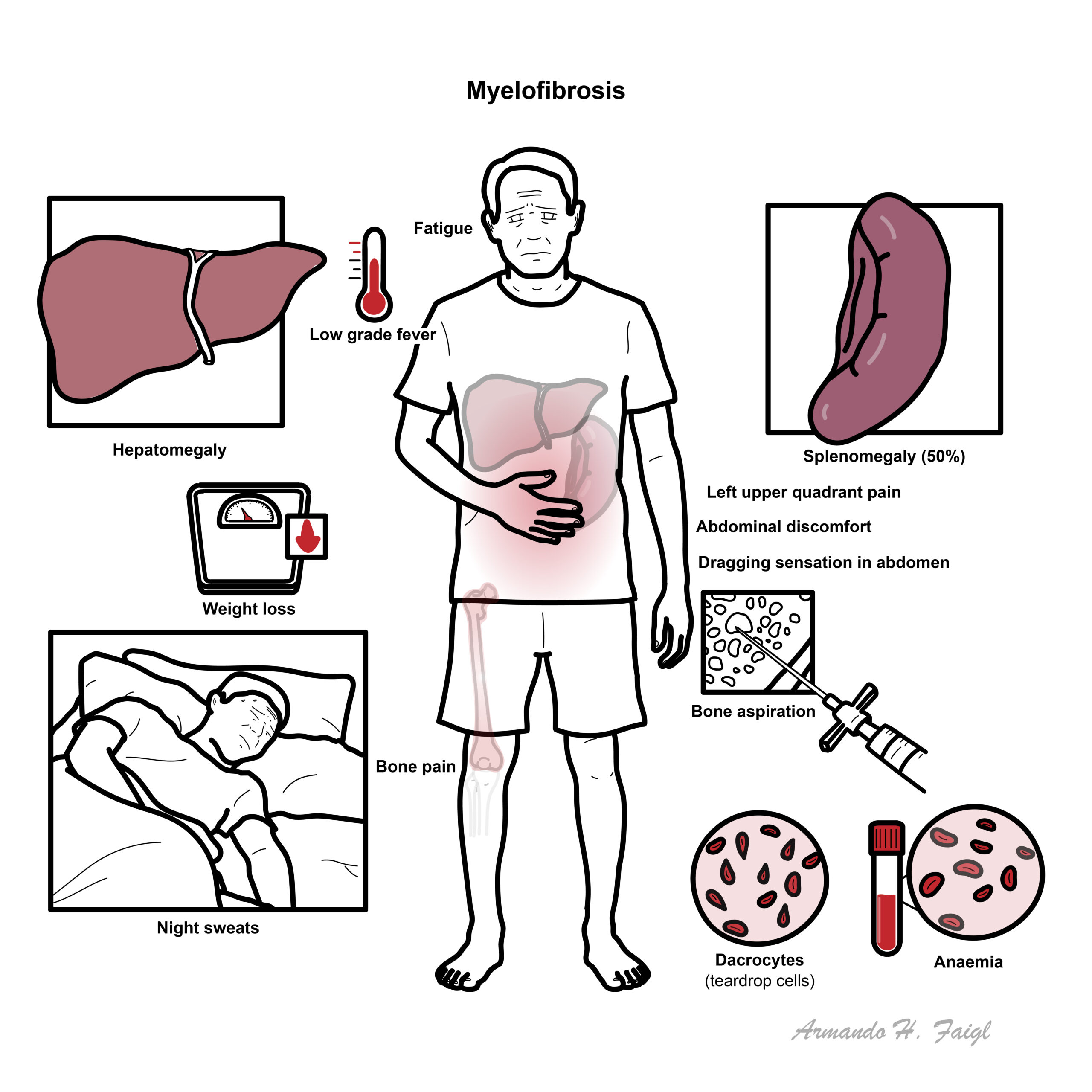
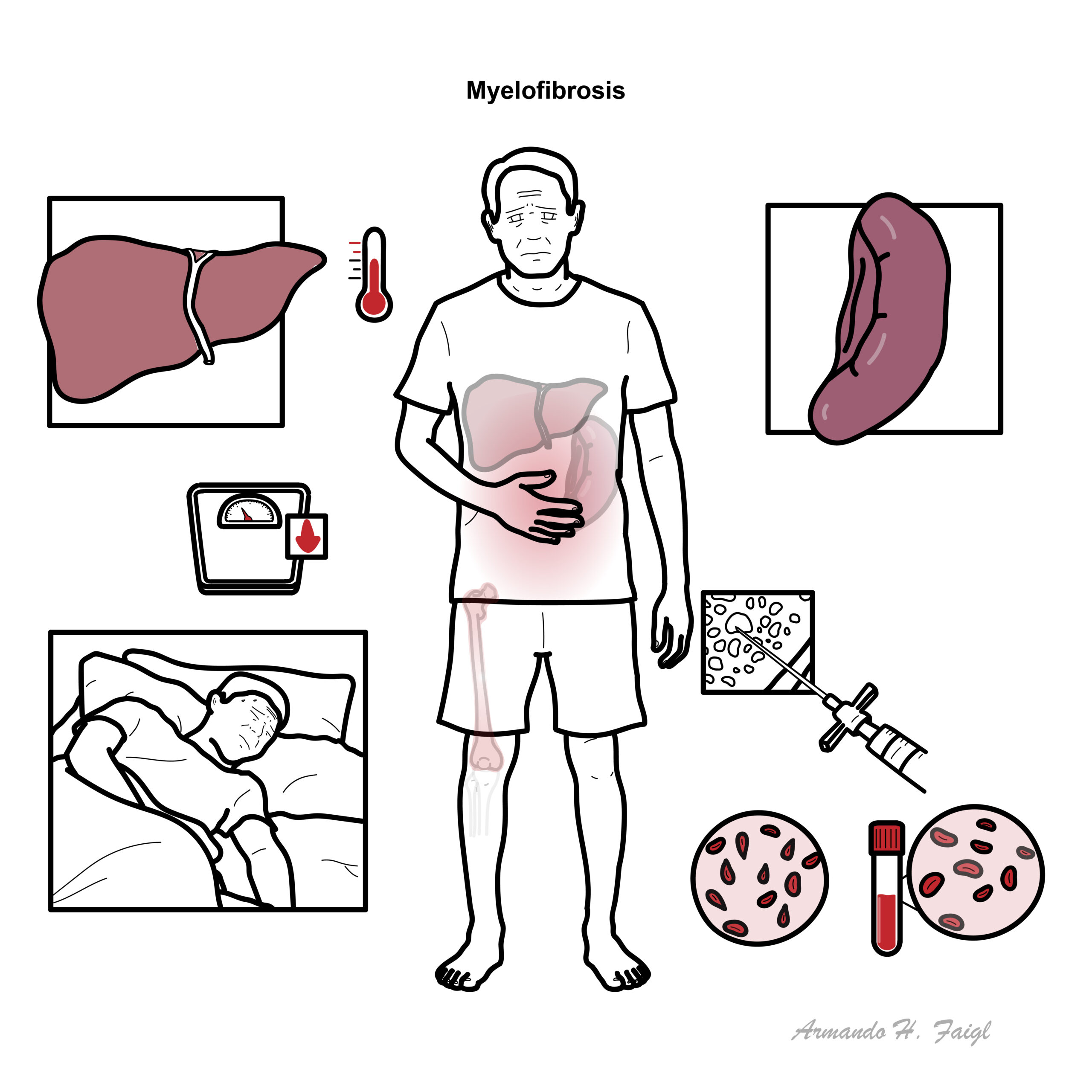
- Fatigue (60%)
- Splenomegaly (50%)
- Hepatomegaly
- Pruritis
Symptoms of splenomegaly (50% of cases)
- Early satiety
- Abdominal discomfort
- LUQ pain
- Dragging sensation in abdomen
- Should tip pain (irritation of the diaphragm)
Symptoms of hypermetabolic state
- Low grade fever
- Night sweats
- Weight loss
- Fatigue
- Bone pain
Sites and signs and symptoms of extramedullary haematopoesis
- Pleura – pleural effusion
- Lungs – pneumoniae and pulmonary hypertension
- Heart – pericardial effusion
- Peritoneum – ascites
- Genitourinary tract – obstruction, dysuria
Investigations
- Full Blood Count – anaemia
- Full Blood Smear – nucleated red blood cells, tear drop red blood cells (dacrocytes)
- EUC – ↑uric acid
- LFT – ↑alkaline phosphatase, ↑lactate dehydrogenase
- Imaging Investigations – CT or MRI
- Bone Biopsy
- Bone Aspiration – dry aspiration
Other tests
- RF, ANA, Anti-CCP
- ↑CD43
Diagnosis The 2008 WHO criteria include a combination of the following findings:
- Presence of megakaryocyte proliferation and atypia, usually accompanied by reticulin and/or collagen
- WHO criteria for polycythemia vera (PV), chronic myeloid leukemia (CML), myelodysplastic syndrome (MDS), or other myeloid neoplasm not met
- Demonstration of a clonal marker (eg, JAK2 or MPL)
- Leukoerythroblastosis
- Palpable splenomegaly
- Anemia
- Increased serum lactate dehydrogenase level
| MAJOR CAUSES OF SPLENOMEGALY | |
| Aetiology | Conditions |
| Congestion | Cirrhosis, Heart Failure, Thrombosis of portal/hepatic/splenic vein |
| Malignancy | Lymphoma, Leukaemia, Myeloproliferative disorders, Primary or secondary tumour |
| Infection | Viral (hepatitis, infectious mononucleosis, cytomegalovirus), bacterial (Salmonella, TB), parasitic (malaria, toxoplasmosis, schistosomiasis), infective endocarditis |
| Inflammation | Sarcoids, SLE, Rheumatoid arthritis (Felty syndrome) |
| Infiltrative, nonmalignant | Gacuher’s disease, Niemann-pick disease, Amyloid, Langerhan cell histiocytosis |
| Haematologic (hypersplenic) states | Haemolytic anaemias, Sick cell disease (children) |
More information on the differential diagnosis of Splenomegaly.
No direct relation between splenic size and hypersplenism, however hypersplenism is more common among those who have gross splenomegaly.
Pathology
- Extensive fibrosis – Collagen and/or reticulin
- Megarkaryote proliferation and hyperplasia
- Immature granulocytosis
Pathophysiology
Hypersplenism is a triad of splenomegaly, pancytopenia and normocellular bone marrow. Splenomegaly increases the spleen’s mechanical filtering and destruction of RBCs and often of WBCs and platelets. Compensatory bone marrow hyperplasia occurs in those cell lines that are reduced in the circulation.
Treatment
Overview
The initial management of patients with PMF is largely dictated by the risk of disease progression and estimated overall survival as calculated by prognostic scores
Asymptomatic (30% of patients)
- Folic acid supplement
- Pyridoxine supplement
- Peginterferon alfa
- Follow up every 6 months with specialist
Symptomatic
- Bone marrow transplant – if suitable
Bone marrow transplant offers the only currative potential for patients with PMF, however only a selected few will be able to have this.
- The JAK2 inhibitor (Ruxolitinib) – not only if JAK2 postitive but can help reduce spleen size.
- Thialidomide + prednisalone if Ruxolitinib is ineffective
- Hydroxyurea – reduce spleen size, control thrombocytosis and leukocytosis, and/or constituional symptoms
- Red cell transfusion – for anaemia
- Danazol +/- erythropoetin – for anaemia
- Allopurinol – for hyperuricaemia
- local irradiation – for the management of symptomatic extramedullary haematopoiesis
- Splenectomy – if other treatment does not work and symptoms can not be controlled
Interferon therapy can be limited by its induction of leukopenia or thrombocytopenia, but it can decrease splenomegaly. This is particularly useful in pregnancy where chemotherapy and thalidomide are contraindicated.
Pharmacology
Ruxolitinib is a JAK2 inhibitor used to treat myeloproliferative disorders. JAK2 (and other kinases) are responsible for the mediation of cytokine and growth factor signalling which in turn effect immune function and hematopoiesis. Side effects anemia, equilibrium disturbance, labyrinthitis, meniere’s disease, dizziness, headache, vertigo, neutropenia, thrombocytopenia, orthostatic dizziness, weight gain and flatulence. No contraindications.
Pharmacology
Danazol is a synthetic steroid (androgen) with antigonadotropic and anti-estrogenic activities that acts as an anterior pituitary suppressant by inhibiting the pituitary output of gonadotropins. Danazol is usually used to treat endometriosis and fibrocystic breast disease (in patients unresponsive to simple measures). Also used for the prophylactic treatment of all types of hereditary angioedema in males and females. Danazol has been shown to stimulate erythropoiesis in patients with refractory anemia, leading to increased Hb level, reticulocytosis, and decreased need for blood transfusions. Side effects of androgen therapy include fluid retention, increased libido, hirsutism, abnormal liver function tests, and hepatic tumors.
Management post Splenectomy
- Prophylaxis for Post-splenectomy infection:
- Vaccinate 2-3 weeks before elective splenectomy: Pneumococcal vaccine, Hemophilus influenza type B (Hib) vaccine, Meningococcal group C vaccine, Influenza vaccine
- Lifelong Antibiotic prophylaxis should be offered in all cases where a patient has an absent or dysfunctional spleen, especially in the first two years after splenectomy, for all children aged up to 16, and when there is underlying impaired immune function: Long-term penicillin 500mg 12 hourly (erythromycin if allergic to penicillin)
- Revaccination of pneumococcal vaccine: in every 5 years and influenza vaccine anually
- Antimalarial chemoprophylaxis: if needed (travel to endemic area)
- General Measures: carrying a “No Spleen” card which details vaccinations, antibiotic therapy and what action should be taken in case of a flu-like illness, advice to seek urgent medical attention at early signs of infection in the future, irrespective of prophylaxis, aspirin prophylaxis against thrombosis is dependent upon the platelet level and should be controlled by a specialist haematologist
- Post splenectomy hematological features:
- Thrombocytosis: persists in 30% cases
- WBC count: usually normal but there may be mild lymphocytosis and monocytosis
- Red cell morphology: Howell-Jolly bodies (Nuclear remnants), Pappenheimer bodies (contain sideroblastic granules), Heinz bodies (Denatured hemoglobin), Target cells, Nucleated erythrocyte (occasionally)
Complications & Prognosis
- Extramedullary involvement
- Anaemia
- Haemorrhage
- Infections
- Complications of splenomegaly
- Postral hypertension
- Leukaemia (20%)
- Hyperuricaemia → gout and renal stone formation
Primary myelofibrosis has a median lifespain of ~5.5 years. Death is usually a consequence of bone marrow failure (haemorrhage, anaemia, or infection), transformation to acute leukaemia, portal HTN, heart failure, cachexia, or myeloid metaplasia with organ failure.




Members only discussions coming soon…